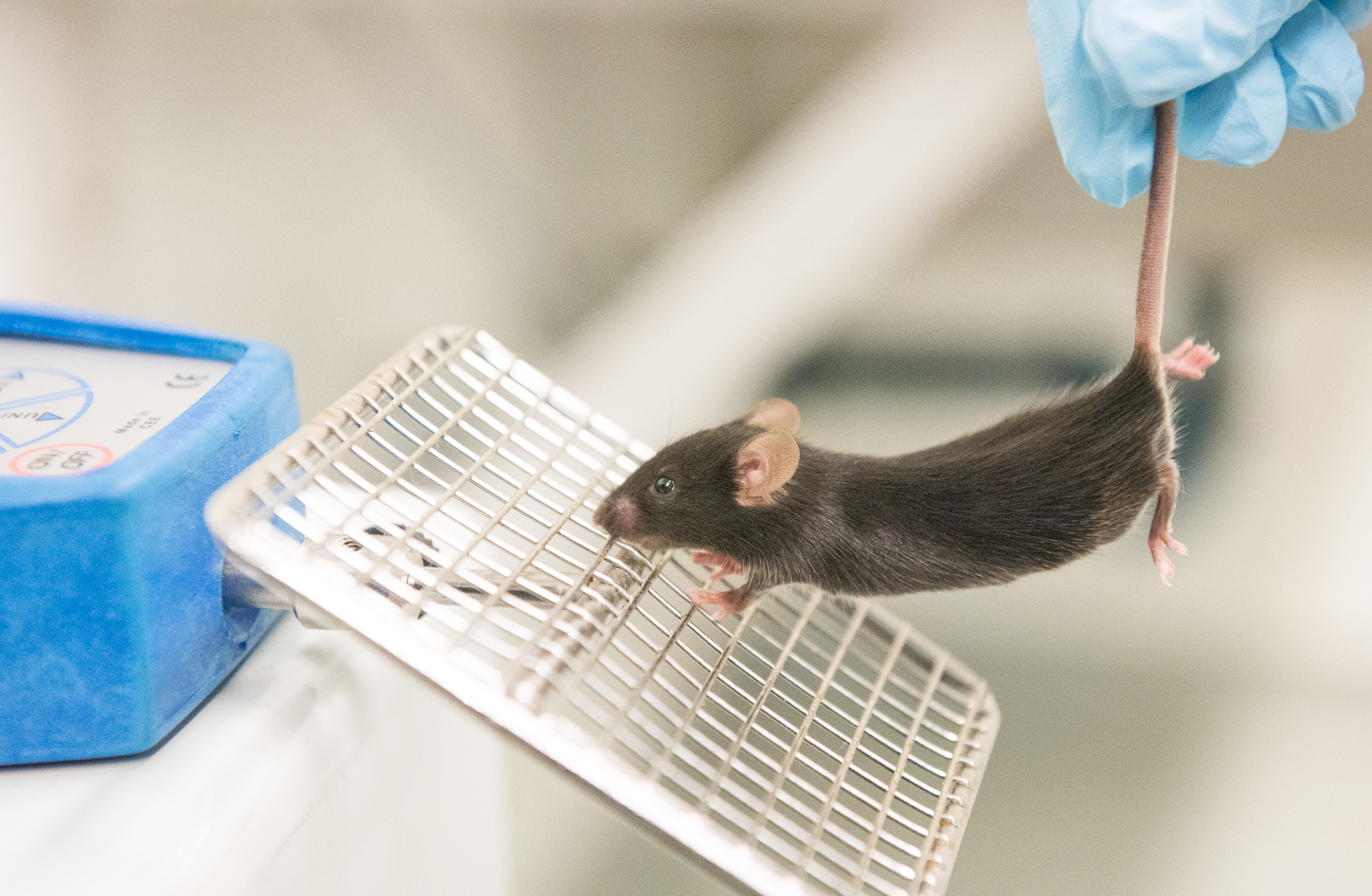
Science and art may feel like completely separate departments but for research scientist Juliette Baker, this couldn’t be further than the truth.
Her mom is a graphic designer and fueled her passion for art while Baker’s own love for science fused the two worlds together. She even drew a diagram for her master’s thesis on Amyotrophic Lateral Sclerosis (ALS).
“Honestly, people don’t think science and art overlap a ton,” Baker said. “But, I’m making tons of schematics for people to understand, and it’s all my artistic brain, really.”
At one point she considered becoming a science illustrator but views painting and art to be more of a de-stressor and hobby rather than a career path. When it comes to science, Baker wants to be more hands-on and focus on direct patient applicable research. However, her path wasn’t picture clear at the start of her academic career.
Baker — who now works at Bond LSC in the lab of Chris Lorson — earned a bachelor’s degree in health science at Mizzou, and planned on going to med school. She worked six months as a medical assistant but realized shortly after that this career path wasn’t for her.
“I love science but what I loved was the research part,” Baker said. “So, I did my masters in a year and a half instead of two years to get back on track.”
After she received her master’s degree in biomedical science last year, an opportunity arose to work at Bond LSC, and she couldn’t refuse.
Baker explained that she had to find three people to comprise a committee for her master’s program. This committee would approve and decide if she would receive her degree. Baker added a fourth person, Chris Lorson, since her research project for her master’s thesis was a collaboration with him and her mentor, Nicole Nichols.
“He [Lorson] emailed me the day I got approved, I think it was before I even found out that I got approved, and said ‘so are you looking for a job?’”
Lorson, a principal investigator at Bond LSC, co-founded Shift Pharmaceuticals to focus on treatment for patients with spinal muscular atrophy (SMA), an inherited motor neuron disease. He was looking for a full-time researcher and Baker checked every box in the job description.
“They were looking for a full-time researcher and it was basically doing exactly what I learned to do my entire master’s so he thought it would be perfect if I could transition to working in his lab,” Baker said.
Baker primarily works for SHIFT but also helps with ongoing projects and aids other students in the Lorson lab.
“It was perfect because I was super worried about having to apply to jobs and figure out what I’m now qualified to do,” Baker said.
Currently, her research is on gene therapy treatments for patients with Charcot-Marie-Tooth disease (CMT). This inherited nerve disease causes abnormalities in the nerves in feet, hands, legs, and arms. She also helps with the SMA projects and SMARD1 project, a rare type of spinal muscular atrophy.
“Honestly this lab helped me realize that [gene therapy] is the kind of specialty that I want to stay in,” Baker said.
However, there is a stigma surrounding gene therapy and people are apprehensive when it comes to this type of treatment. One common misconception is that scientists are altering DNA or changing body composition, which Baker explains isn’t the case.
“What we are trying to do is to modify the production of the gene so it will go back to normal levels,” Baker said. “So, it’s not making the gene any different or adding a new gene that wasn’t there before, we are just modifying how your body is producing [the gene].”
Baker hopes the stigma surrounding gene therapy decreases over time as more people use it for different diseases, and it’ll become more accessible. The more attention this type of therapy receives, the cheaper it will become for people in the future.
“That’s the biggest thing that I hate about my job is that, yes, this treatment is going to be amazing and lifesaving, but it’s going to be expensive,” she said.
She plans on going back to school and getting her Ph.D. and one day working in the pharmaceutical industry. But for now, she enjoys working with Lorson and other undergraduates in the lab.
“The most rewarding part is definitely getting to see how my work and everything I do every day directly affects people with the diseases,” Baker said.
Haval Shirwan and Esma Yolcu arrive at Bond LSC as accomplished researchers.
While having different expertise within the field of immunology, the married couple collaborates extensively on research and together developed ProtEx technology, an alternative to traditional methods of gene therapy for immunomodulation with applications to autoimmune diseases, transplantation, cancer immunoprevention and immunotherapy, and vaccines against infections
With work published in numerous peer-reviewed journals and 19 patents to Shirwan’s name, they both arrive at Mizzou ready to build on their research background and contribute to the new precision medicine focus for the UM System.
But success didn’t always come easily for the couple.
Shirwan grew up in the shadows of Mount Ararat in eastern Turkey, and was always fascinated by how the beautiful nature in the area came to life during spring after a harsh winter. From then, he knew he wanted to spend his life studying something in a science-related field.
After completing his undergraduate education in Turkey, Shirwan received a scholarship via NATO to complete his Ph.D. in the United States at UC-Santa Barbara. From there, he obtained a postdoctoral fellowship at California Institute of Technology, where he worked under an esteemed group of researchers, spent a few years in Philadelphia and arrived at the University of Louisville where he has spent over 20 years in the Department of Microbiology and Immunology.
Yolcu also grew up in Turkey, but did not meet the man who is now her husband until much later in life.
After completing her undergrad and Ph.D. in her homeland, she faced a difficult decision, between continuing in her field of training — cancer biology — or taking a leap of faith to pursue her emerging passion of immunology. That decision would require her to move to the United States for a fellowship at the University of Louisville, which she did. She originally planned to return to Turkey after the fellowship, but after meeting Shirwan, decided to settle in the US and make her career here.
Shirwan and Yolcu bonded over their passion for immunology worked together to develop their esteemed ProtEx technology. But, their initial desire to do so came not out of inspiration but rather out of frustration at the shortcomings of traditional gene therapy methods.
“DNA-based gene therapy is a complicated, exhaustive and expensive technology,” Shirwan said. “We wanted to see if there was a more efficient way to do immunomodulation.”
In traditional methods of gene therapy, DNA must first be introduced into the cell, but ProtEx technology bypasses that step, instead sticking the protein directly on the cell surface for immunotherapy.
The advantages of this method are twofold: it is much more efficient than gene therapy and allows proteins which may be harmful to the cell to “get out of the way” quicker once the process is completed.
Although this technology has received much acclaim from the global scientific community, the breakthroughs took years of work. While Shirwan took charge of the more technical details of ProtEx, it was Yolcu who urged him to be patient and keep trying new strategies when he was ready to give up on the technology altogether.
“In the mid 90s was when we had our breakthrough with ProtEx,” he said. “By the time we reached that point, I was about ready to give up, but (Yolcu) urged me to keep trying.”
Their breakthrough came when a group of animals treated with ProtEx techniques experienced unprecedented recession of cancerous tumors on their body. At first skeptical, Shirwan and Yolcu repeated the experiment several times. The result was the same; the control groups saw enlarged tumors while the group treated with ProtEx technology saw their tumors recede greatly. Their innovative immunomodulation technique of placing protein directly on the cell surface had worked. The couple published their ProtEx findings initially in 2002, but continued to fine-tune ProtEx and discover new things along the way.
While they have already accomplished so much together, they believe they still have much left to learn.
“What’s so fascinating about immunology is that we often think we’ve made so much progress and know so much about the immune system, but in reality, we still know very little,” Yolcu said. “Studying immunology gives me a sense of peace, and I’m excited to continue our work at Bond LSC.”
To that end, the couple aims to establish themselves at Mizzou as soon as it’s safe to do so (they are currently in the midst of a multi-phased moving process due to COVID-19).
This type of research contributes to the University of Missouri System’s NextGen Precision Health focus. The NextGen Initiative unites scientists, government and industry leaders with innovators from across the system’s four research universities in pursuit of life-changing precision health advancements.
Shirwan and Yolcu will establish a Center for Immunomodulation and Translational Research and aim to collaborate with clinical colleagues and scientists from all different backgrounds to continue to make headway in their work with the immune system.
“We want to build a powerhouse here,” Shirwan said.
For scientists, studying a disease presents a puzzle looking for an answer, but there are real people behind the research that may one day cure the illnesses that turned their lives upside down. Chris Lorson and Monir Shababi work on one of these puzzles in Bond LSC.
Find out more about their work and the faces behind SMARD, a rare, often fatal, genetic motor neuron disease in the following story courtesy of the College of Veterinary Medicine.
Monir Shababi, an assistant research professor in veterinary pathobiology, and Christian Lorson, Bond LSC principal investigator, College of Veterinary Medicine professor and associate dean for research and graduate studies, have invested countless hours during the past five years to solving a cruel medical mystery. A family who has endured the agonizing ordeal of having two children born with the same disease has invested funding for the research being conducted at MU’s Bond Life Sciences Center.
Meet the Sims family: mother, Jill Sims, MD; daughter, Caroline; father, Eric, an associate professor of economics at Notre Dame; daughter, Molly; and daughter, Catherine, who turned 5 in August. Catherine is living with a rare genetic disorder called Spinal Muscular Atrophy with Respiratory Distress, or SMARD. The Sims had another child, Bobby, who was born in 2012 and died of SMARD after a month of life.
The disease is called spinal muscle atrophy with respiratory distress, or SMARD. SMARD is a progressive motor neuron disease that has no treatment or cure. At least, not yet.
Shababi, PhD, and Lorson, PhD, and the Sims family — mother Jill, father Eric, grandparents Grant and Patricia — have teamed up in an effort to change that.
*****
The disease is so rare that it is largely unknown, even to most medical professionals. When you are the parent of a child with SMARD, you are in a daily, nonstop, life-and-death struggle.
It is exhausting. It is frustrating. It is a battle that requires an endless reserve of endurance and willpower. And, it requires cutting-edge, scientific discoveries that are just coming to light at MU’s Bond Life Sciences Center.
*****
Catherine Sims lives on a ventilator and needs around-the-clock care. Yet, now age 5, her life is a victory.
“Our first daughter was born healthy, so we had no idea that we carried such a terrible disease,” Jill Sims says. “Then, our second child, Bobby, — who is named after my dad — was born very small, which was unusual given our family history, and he was very quiet as an infant. Those were the only things I noticed. He was three weeks old and we were driving back from Thanksgiving at my parents’ house. I fed him and put him in his car seat. I checked on him 30 minutes later and he had died. He had aspirated. The disease causes the diaphragm not to work, so he couldn’t breathe and eat at the same time.”
Despite requiring mechanical ventilation and around-the-clock care, Catherine attends school, enjoys family outings and participates in activities as much as possible.
Bobby Sims, born Oct. 31, 2012, died on Nov. 30, 2012. His death was attributed to unknown respiratory failure, and he was considered a victim of Sudden Infant Death Syndrome (SIDS). Catherine Sims was born in August 2013; her diagnosis came four months later.
“I went on to have Catherine next, and then we knew something was up,” Sims says. “Catherine was very similar to Bobby, very small and very quiet. That, of course, led us to figure out something was going on.
“In the period of time when Catherine was having problems and was hospitalized but undiagnosed, Catherine had a test done that put her group of symptoms into a specific category of neuromuscular diseases,” Sims says. “A good friend of mine Googled that category and the search produced a WordPress blog that Lisa Porter Werner had contributed to.”
The blog contained personal stories of families who had children with a disease named SMARD. The goal of the blog was to put SMARD on the radar, for families who didn’t have a diagnosis and needed to find answers as well as find support. Porter Werner had posted her own family’s story.
“My friend forwarded me Lisa’s particular story regarding her two children with SMARD, and the story almost identically matched my own,” Sims recalls.
Porter had read extensively and combed the internet for information and cases similar to those of her children. Porter eventually found a modicum of information about something called SMARD, which had been diagnosed in approximately 60 children.
“Lisa Porter’s blog contained the personal stories of families who had children with SMARD,” Sims recalls. “My friend forwarded me Lisa’s particular story regarding her two children with SMARD, and the story almost identically matched my own.
Living with SMARD, a progressive motor neuron disease, means Catherine needs 24/7 care from her family and home-health attendants.
“The Werner’s first daughter died at six weeks of age. It was called a SIDS case; she just died in her sleep,” Sims says. “They had Silas, their son who is living with SMARD, shortly thereafter and she put him in a sleep study when he was three weeks old. She said, ‘No, my daughter didn’t just die. There was a reason.’ It turned out that Silas was having major breathing problems during sleep.
“I was convinced after reading about Lisa’s family that my two children had SMARD as well, and I asked Catherine’s doctors to test her for it,” Sims says. “Catherine’s test came back positive four weeks later. A year or so later, I connected with Lisa through a Facebook group for families with children with SMARD. We began talking more, once my in-laws funded SMARD research at the Jackson Lab, and continued to talk once we found out about Dr. Shababi’s paper that came out in 2016.”
*****
In order to know what SMARD is, it is important to know what it is not. Despite the obvious similarities in name, spinal muscular atrophy (SMA) and spinal muscular atrophy with respiratory distress have sharp differences.
Both conditions affect the lower motor neuron cells of the spinal cord that control voluntary muscle activities like walking, talking, breathing and swallowing. Both are sometimes characterized as “like ALS in babies.”
SMA, which can range from type 1-4, is caused by mutations in or the absence of the SMN1 gene. SMA typically causes weakness in the core first and the baby or child may present as hypotonic, or having low muscle tone — sometimes called floppy baby syndrome. Babies or children with SMA may eventually develop respiratory compromise over time.
SMA is the leading genetic killer of infants; one in 40 people are carriers of SMA.
SMARD, in contrast, is extremely rare. The exact number of cases is unknown, but it has clearly occurred in more than the approximately 100 children worldwide who now carry that tragic diagnosis. SMARD is branded an “orphan” disease, a term commonly applied to any debilitating medical condition that affects fewer than 200,000 Americans. There is little information and few resources available regarding SMARD.
The Sims sisters: Catherine, a SMARD survivor, with older sister Molly and younger sister Caroline. Catherine is standing with the aid of a device.
SMARD is a genetic disease, caused by mutations or loss of the IGHMBP2 gene, Immunoglobulin MU-binding protein 2. The condition is inherited in a recessive pattern, meaning both parents must be carriers of the gene mutation and each parent must pass along a copy of the mutation in order for the child to be affected. In essence, every time two carriers have a baby, there is a one in four chance their child will be affected.
Onset of the disease usually occurs suddenly, in what seems to be an otherwise healthy baby, typically between 6 weeks and 6 months of age. Once the diaphragm is paralyzed, the infant must depend on their accessory muscles to breathe. These muscles also weaken as the disease progresses, until the child needs mechanical ventilation.
Many children die in the first year of life, often in their sleep or from a respiratory illness. Past the age of 1 year, almost all children living with SMARD require a tracheostomy, a ventilator and a wheelchair.
Simply put, SMA usually presents as a hypotonic or “floppy” baby who gradually develops respiratory distress. SMARD presents as a baby in respiratory distress who gradually becomes hypotonic.
SMA and SMARD share a similarity in that both are monogenic disorders, conditions caused by mutations or loss of a single gene. Shababi and Lorson have an established history of working with SMA. Now, their focus is SMARD.
*****
“In 2009 and 2010, a lab at the Ohio State University used a viral vector to introduce the SMN gene in SMA mice,” Shababi, the CVM researcher, says. “The viral vector does not contain the necessary genes required for the virus to cause infectious disease. You can replace viral genes with the specific gene you want and keep only the part of the virus that is required to enter the body, find its receptor and produce the desired protein from the gene it carries.
“They (researchers at Ohio State) put a human SMN gene into a viral vector — adeno-associated virus 9 (AAV9) — that has the potential to pass the blood brain barrier in humans. This virus has the capability to enter into the brain, the spinal cord, muscles and peripheral organs,” Shababi continues. “The AAV9 virus carrying the SMN gene was injected into SMA mice. They were able to rescue the affected mice. That was a huge step toward treating SMA. That vector is currently in Phase 2 clinical trials with AveXis/Novartis.
“With SMARD, there is also a single gene involved in the disease — the IGHMBP2 gene,” Shababi continues. “So, we took a human IGHMBP2 gene, in the form of cDNA, and placed it into the same AAV9 vector and injected it into the brain of SMARD pups that were 2 days of age. Our virus did the job and the SMARD mice were cured.”
*****
Catherine and older sister Molly sporting festival face paint.
“Dr. Shababi posted a paper, I believe in March 2016, that reported the results of her work on SMARD,” Sims says. “Lisa found the paper and contacted Dr. Shababi and had a wonderful reception. They had several very long conversations about what Monir was doing, what she had already been doing, and they immediately had a strong connection.
“Dr. Shababi was very personable over the phone, and was very passionate and very approachable about her work,” Sims relates. “Sometimes, it’s hard to get ahold of people, but Monir answers her own phone, and she was very clear with Lisa about what had already been done, which was pretty cool for us because we didn’t know — we didn’t realize how much work Dr. Shababi and Dr. Lorson had already done on SMARD. We were impressed by how much of a handle they already had on the disease. They were ahead of the game. That was great news for us on the family side; at the time, we were aware of only one other lab in the country — the Jackson Lab in Maine — doing work in this area. We couldn’t believe that, wow, there’s a second lab and they are already in gear, they already have a lot of good things going.
“Then, Lisa got me in the loop with Monir, and I talked to her a few times,” Sims continues. “They were having a funding issue, which is not surprising because of how rare the disease is. When we first learned about the work being done at the Jackson Lab, my in-laws agreed to fund SMARD research at Jackson. After learning what Dr. Shababi and Dr. Lorson were doing, I talked to my in-laws again and asked if they would be interested in funding Monir’s research. My father-in-law and I had a few conversations with Monir and Chris Lorson, and then my in-laws decided to do another fund, this time at Missouri, that started this past December.”
*****
“If you look back a number of years, there has been a gene therapy on the translational side that has had exceptionally powerful results in SMA,” says Lorson. “AveXis now has a Phase 2 clinical trial going for their gene therapy product, which has the potential to be very impactful. It has demonstrated efficacy in SMA, but also provides an important proof of principle for gene therapy as a whole. So, it was really exciting to know that there’s only one gene responsible for each of these horribly devastating diseases, SMA and SMARD. It allows you to consider following a similar path. Knowing that, Monir started developing a project that was gene therapy, gene replacement for SMARD.
“Whenever I talk about this, I give about 110 percent of the credit to Monir,” Lorson explains. “Monir has really been the driver of this entire project. Originally, I said, ‘Monir, I’d really like you to develop this gene therapy for SMARD, I think it’s a really exciting area of research. I’ll check back in about six months.’ When I did, we had the mice, we had the vector and she was doing the experiments. That’s exactly the kind of gumption that you hope to find. She did all of that. My role was to say, ‘Good job, Monir!’
“She was the first author on an important paper in Molecular Therapy published in 2016,” Lorson continues. “Based upon that, and the level of excitement, people found her. Through Facebook and Facebook friends, they started to communicate back and forth. Monir is driving it. Monir is doing it.
“AAV9 is in clinic for a number of other diseases, but every time you put a new gene in, you have to go through the Food and Drug Administration,” Lorson says. “That’s why the process isn’t as simple as it might appear to be. Every single time you change that vector — that gene delivery vehicle — you have to get it approved.”
*****
“My in-laws have been very generous, but you need a lot of capital to do this research,” Jill Sims says. “SMARD is so rare that progress will probably come only from academic research. You really need a lot of support and you need a lot of funding from various sources. Right now, our life continues the same. It’s great that everybody is doing this great research, but you need so much more for a cure. That’s what everybody wants; we want our kids to be normal.
“A day in the life of someone with SMARD is very difficult,” Sims says. “There’s a lot that has to be done to have a normal life, and there are a lot of obstacles to that, so you’re constantly trying to overcome those.
“This disease is devastating,” Sims continues. “It can take away every basic human function: the ability to sit, crawl, stand, walk, talk, swallow, feed oneself, clean oneself, use writing utensils and so on. The disease also makes the person more likely to have respiratory problems since they can’t breathe or even cough on their own. It is hard as a parent. Every day we live with the potential fatality of this disease. If their trach tubes come out, they cannot breathe. These trachs sit in their windpipes, held in by ties, like a tight necklace. It is not secure.
“You may go months without anything happening then, all of a sudden, it’s coming out. When that happens, she may only have 60 or so seconds to live,” Sims says. ”You have to have someone always watching them, either a specially trained nurse or a parent, who is a trained caregiver.
“That’s the hard part that we always live with,” says Sims. “Yes, she looks good, and she goes to school, and she’s in activities, to some degree. We adapt everything so she can do as much as possible. But, she is living with a fatal disease that is non-treatable. We basically just manage her symptoms. We know very well that we could lose a second child. That’s what is hardest on us. Even though there are these great advances, she is alive because of amazing machines. Every day presents the chance that she could die.
“When we take Catherine places, there are always at least 10 machines that go with her,” Sims says. “Everything just takes longer. We have a special van with a lift, because she’s in a wheelchair. You are in the thick of trying to make what is not normal to be normal.
“You can’t just pick up your child and go, you can’t feed them a different way, or put a different outfit on them,” Sims continues. “Those are the silly things I took for granted having had a healthy child before. I just did her hair, brushed her teeth, and put her in whatever, and fed her whatever I wanted. Catherine cannot do that. It’s the small things that you take for granted, and there are so many ‘small’ things. We are fortunate to have excellent in-home nursing care, but this also means that my husband and I have had to sacrifice a lot of our privacy. And, I’ve had to give up a lot of my mothering, because I have someone else that always needs to know what I’m doing. That’s hard.
“So, we want a cure,” Sims states. “We are all in. We are always fighting the disease. Our goal would be to have a cure as fast as possible, because the older the kids get, the less chance you have of curing them. This is a neurologic disease; it is hard to get those nerves back. We realize that our kids may be too old. Catherine will be 5; Lisa’s Silas is 8 or 9. They’re kind of old. The ideal time would be right at birth or shortly thereafter. So, that’s what we want. We want the big places — the big funding sources — to realize how important this is, even though it affects only a small number of people.”
*****
“Our gene therapy vector is a very powerful tool,” Lorson says. “It is early days, in terms of trying to push it to the clinic, but we’re trying to do all the important pre-clinical questions.
“There are a number of questions you have to ask,” Lorson continues. “When do you deliver that kind of vector? Does it work only if you do it right at birth, before disease develops? Can you correct the disease, in other words, once the research animals have the disease, can you bring them back to more of a normal state? Or, once that happens, is it just too late for something like gene therapy? We want to deliver what they want to see, in terms of working hard and getting results out. That is what we are trying to do.
“I want to say, ‘Thank you,’ in the biggest way possible to the Sims family,” Lorson says. “Their generosity is really amazing. We consider this an exceptional honor. We want to be the best stewards they could possibly find, of their trust and of their funds. People go out and raise these funds — in some cases, through car washes and bake sales — so you have to put a particularly high value on those dollars. My fondest hope is that we do that every day.”
If you would like to help in the battle against diseases that could someday be relieved through gene therapy, please visit this page.
Kevin Kaifer, a Ph.D candidate who works in Dr. Christian Lorson’s lab. | Photo by Mary Jane Rogers, Bond LSC
By Mary Jane Rogers | Bond LSC
“#IAmScience because there are people suffering all over the world and this is where I’m most likely to make any kind of an impact.”
When he came to MU three years ago, Kevin Kaifer knew he wanted to work in Bond LSC. He felt it was where the best science and collaborations were happening on campus, and everything that he needed for his research – a vivarium, a Genomics Technology core, and proteomics core – were all conveniently housed here.
“I entered research because I thought the complexity of cellular life is the most fascinating topic in the world,” said Kaifer. “I wanted to be a part of it.”
He completed his undergraduate degree in biology at Truman State University and is currently part of Dr. Christian Lorson’s lab. There, Kaifer is learning transferable skills – everything from communication skills to the production of recombinant gene therapy vectors – all of which will give him a strong foundation for a career in industry.
“The growing promise of gene therapy as a safe and realistic treatment option has led to the start up of many biotech companies that are making really exciting progress,” he said. “This is where I think I will be best able to contribute to science and therapy.”
For undergraduate students who are just getting started in a science field, Kaifer emphasizes that success in science comes and goes.
“In my own personal experience, success in science only comes after a significant set of hurdles,” he said. “You have to be okay with feeling stupid, because part of your job description is to answer questions to which you do not know the answer. I would actually be concerned if you were not struggling to feel successful.”
Gene therapy treating the neurodegenerative disease, SMARD1, shows promising results in mice studies.
Shababi uses an instrument to measure grip strength in the forelimbs of mice. Healthy mice are able to cling on with a stronger grip than SMARD1 mice. | photo by Jennifer Lu, Bond LSC
Monir Shababi was confident her experiments treating a rare genetic disease would yield positive results before she even ran them.
Scientists had success with a similar degenerative neuromuscular disease, so she had every expectation their strategy would work just as well in her mice.
Monir Shababi, an assistant research professor in the Department of Veterinary Pathobiology, studies SMARD1 in mice. | photo courtesy of the Department of Veterinary Pathobiology
“I was expecting to get the same results,” said Shababi, an assistant research profession in Christian Lorson’s lab at the University of Missouri Bond Life Sciences Center. Shababi studies spinal muscular atrophy with respiratory disease type 1, or SMARD1.
The treatment worked, but not without a few surprises.
Her findings, published in Molecular Therapy, a journal by Nature Publishing Group, are one of the first to show how gene therapy can effectively reverse SMARD1 symptoms in mice.
In patients, SMARD1 is considered such a rare genetic disorder by the U.S. National Library of Medicine that no one knows how frequently the disease occurs. It’s only when babies develop the first symptom—trouble breathing–that pediatricians screen for SMARD1.
Shortly after diagnosis, muscle weakness appears in the hands and feet before spreading inwards to the rest of the body. The average life expectancy for a child diagnosed with SMARD1 is 13 months. There is currently no effective treatment.
Since the neuromuscular disease is caused by a recessive gene, SMARD1 comes as a shock to the parents, who are carriers but do not show signs of the illness, Shababi said. This genetic defect prevents cells from making a particular protein that scientists suspect is vital to replication and protein production.
The hereditary nature of the disease has a silver lining, though. Because SMARD1 is a caused by a single pair of faulty genes and not multiple ones, it is a prime candidate for gene therapy that could restore the missing protein and reverse the disease.
To do that, Shababi set up a dose-response study using a tiny virus to carry the genetic instructions for making the missing protein. She injected newborn mice with a low dose of the virus, a high dose, or a placebo with no virus at all.
Injecting at different doses allowed her to ask which dose worked better, Shababi said.
According to the previous research, a higher dose should have resulted in a more effective treatment.
“So I thought a higher dose was going to work better,” Shababi said.
Instead, the high dose had a toxic effect. Mice given more of the virus died sooner than untreated mice. Meanwhile, mice given a low dose of the gene therapy lived longest. They regained muscle function and strength in both the forearms and the hind limbs and became more active.
In fact, some of them survived long enough to mate and produce offspring.
Initially, Shababi housed her SMARD1 mice in the same cage as their mothers so that the moms could intervene if the sick pups become too feeble to feed themselves. When the male pups became well, their moms became pregnant.
“That was another surprise,” Shababi said. “That was when I knew I had to separate them.”
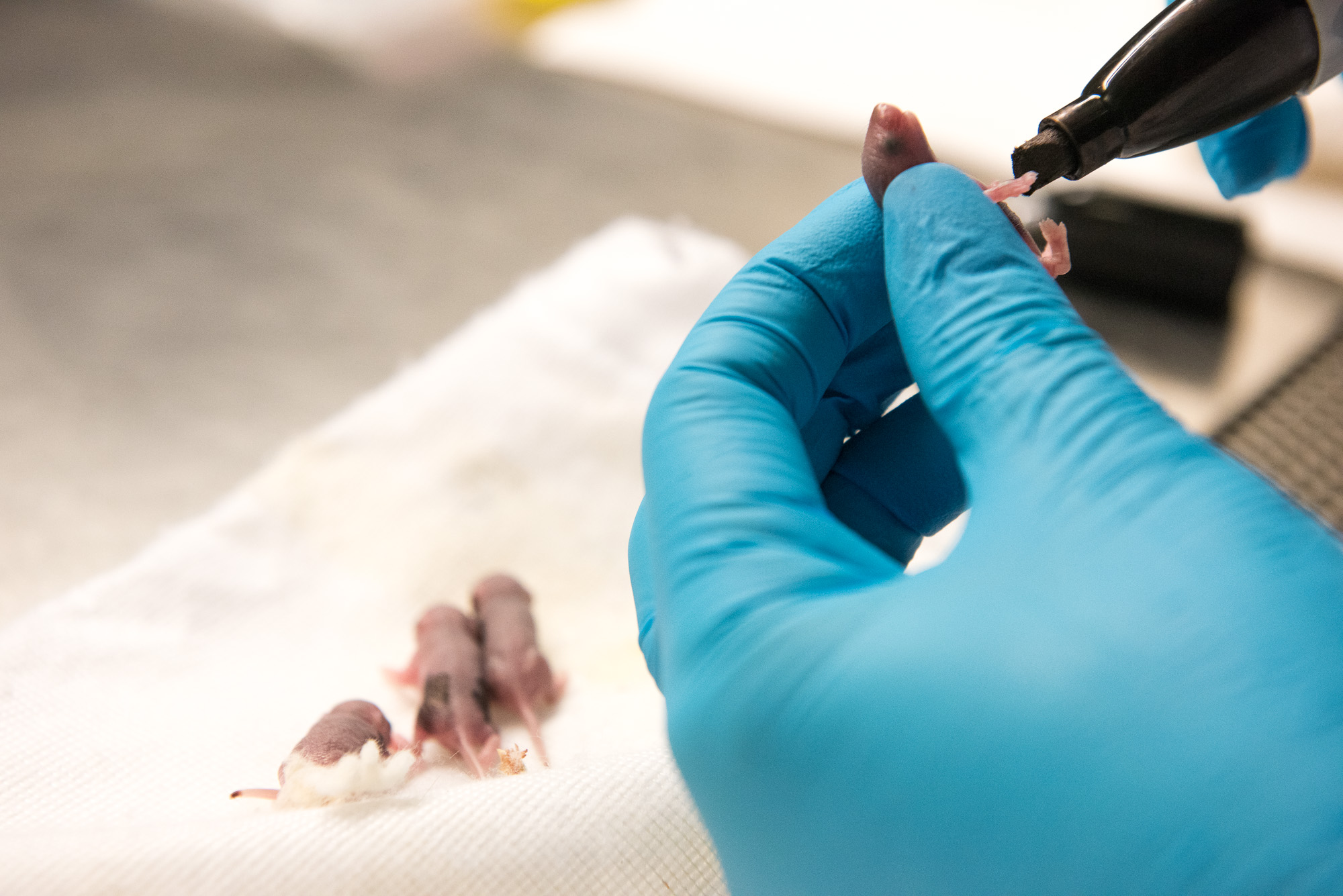
Shababi marks a pup, only a few days old, with permanent marker so each mouse in her study can be identified. | photo by Jennifer Lu, Bond LSC .
In another twist, Shababi discovered that the route of injection also mattered.
To get the treatment across the blood-brain barrier and to the spinal cord, Shababi used a special type of injection that passes through the skull and the ventricles of the brain, and into the spine.
This was no easy task.
The newborn mice were no larger than a gummy bear. To perform the delicate work, Shababi — who has written a chapter in a gene delivery textbook about this procedure — had to craft special needles with tips fine enough for this injection. She added food coloring to the injection solution so she could tell when it had reached its intended destination.
“After half an hour, you will see it in the spinal cord,” Shababi said. “The blue line in the spine: that’s how you can monitor the accuracy of the injection.”
Unfortunately, repeated injections in the mice caused hydrocephaly, or swelling in the brain.
“They get a dome-shaped head,” Shababi explained.
The swelling happened in all three treatment groups, but most frequently in the group that received a high dose of viral gene therapy. This reinforced the finding that while a low dose was beneficial, a high dose was even more harmful than no treatment at all. It’s unclear why.
Christian Lorson is a professor of veterinary pathobiology at the Bond LSC. His research focuses on spinal muscular atrophy and more recently, SMARD1. | photo by Hannah Baldwin, Bond LSC .
The Lorson lab plans to continue studying SMARD1 and this treatment, in particular, how changing the delivery routes for gene therapy can improve outcomes in treating SMARD1.
“It’s not as simple as replacing the gene,” Lorson said. “It comes down to the delivery.”
Injections in the brains of mice are meant to mimic spinal cord injections in humans, but intravenous delivery could be another option. However, intravenous injections, which travel through the blood stream and to the entire body, might cause off-target effects that could interfere with the effectiveness of the treatment.
Once researchers better understand how to optimize dosing and delivery on the cellular and organismal level, the therapy can move closer to clinical trials, Lorson said.
Even though gene therapy for SMARD1 is still in its early stages, he said he was optimistic that developing treatments for rare genetic diseases is no longer the impossible task it seemed even ten years ago.
Spinal muscular atrophy (SMA) is a prime example of a recent success, Lorson pointed out. In the last six years, gene therapy for that disease has moved from the research lab to Phase I clinical trials.
“While it feels like a long time for any patient and their families,” Lorson reassured, “things are moving at a breakneck pace.”
The study, “Rescue of a Mouse Model of Spinal Muscular Atrophy With Respiratory Distress Type 1 by AAV9-IGHMBP2 Is Dose Dependent,” was published in Molecular Therapy, a journal published by Nature Publishing Group. This work was supported by a MU Research Board Grant (C.L.L.); MU College of Veterinary Medicine Faculty Research Grant (M.S.); the SMA Foundation (C.P.K.); National Institute of Health/National Institute of Neurological Disorders and Stroke grants; and the Missouri Spinal Cord Injury Research Program (M.L.G.).
Graduate students Yuleam Song and Dan Salamango inoculate a bacteria culture in Johnson’s lab. The inoculation takes a small portion of a virus and multiplies the sample, allowing researchers to custom-make viruses.
By Madison Knapp | Bond Life Sciences Center summer intern
Modern science has found a way to turn viruses —tiny, dangerous weapons responsible for runny noses, crippling stomach pains and worldwide epidemics such as AIDS— into a tool.
Gene therapy centers on the idea that scientists can hijack viruses and use them as vehicles to deliver DNA to organs in the body that are missing important genes, but the understanding of virus behavior is far from exhaustive.
Marc Johnson, researcher at the Christopher S. Bond Life Sciences Center and associate professor of molecular microbiology and immunology in the MU School of Medicine, has been building an understanding of viral navigation mechanisms which allow a virus to recognize the kind of cell it can infect.
Johnson’s research specifically explores the intricacies of the viral navigation system and could improve future direction of gene therapy, he said.
Marc Johnson (left) with Dan Salamango, a graduate student that works in his lab. The lab does important research on the basic function and mechanisms of viral navigation and transport.
Turning a virus into a tool
Conceptualized in the 1970s, gene therapy was developed to treat patients for a variety of diseases, including Parkinson’s, leukemia and hemophilia (a genetic condition that stops blood from clotting).
To treat disease using gene therapy, a customized virus is prepared. A virus can be thought of as a missile with a navigation system and two other basic subunits: A capsule that holds the ammunition and the ammunition itself.
The viral genetic material can be thought of as the missile’s ammunition. When a cell is infected, this genetic material is deployed and incorporated into the cell’s DNA. The host cell then becomes a factory producing parts of the virus. Those parts assemble inside the cell to make a new virus, which then leaves the cell to infect another.
The capsule is made of structural protein that contains the genetic material, and the navigation system is a protein that allows the virus to recognize the kind of cell it can infect.
Viral navigation
Gene therapy uses viruses to solve many problems by utilizing a virus’ ability to integrate itself into a host cell’s DNA; to do this successfully, researchers need to provide a compatible navigation component.
In the body, viruses speed around as if on a busy highway. Each virus has a navigation system telling it which cells to infect. But sometimes if a virus picks up the wrong type of navigation system, it doesn’t know where to go at all.
“What you can do is find a virus that infects the liver already, steal its navigation protein and use that to assemble the virus you want to deliver the gene the liver needs,” Johnson said. “You can basically take the guidance system off of one and stick it onto another to custom design your virus.”
But this doesn’t always work because of incompatibility among certain viruses, he said.
Johnson and his lab are working to understand what makes switching out navigation proteins possible and why some viruses’ navigation systems are incompatible with other viruses.
“I’m trying to understand what makes it compatible so that hopefully down the road we can intelligently make others compatible,” Johnson said.
The right map, the right destination
Johnson creates custom viruses by introducing the three viral components—structural protein, genetic material, and navigation protein—to a cell culture. The structural protein and genetic material match, but the navigation component is the wild card. It could either take to the other parts to produce an infectious virus, or it could be incompatible.
Johnson uses a special fluorescent microscope to identify which viruses assembled correctly and which didn’t.
A successful pairing is like making a match. If a navigation protein is programmed to target liver cells, it’s considered a successful pairing when the virus arrives at the liver cell target location.
The scope of gene therapy continues to widen. Improved mechanisms for gene therapy, and greater knowledge of how a navigation protein drives a virus could help more people benefit from the vehicles viruses can become.
Johnson uses several high-profile model retroviruses, including human immunodeficiency virus (HIV), which affects an estimated 35 million people worldwide each year, according to the World Health Organization.
Understanding nuances of HIV in comparison to other viruses allows Johnson to pick out which behaviors might be common to all retroviruses and others behaviors that might be specific to each virus.
Johnson said his more general approach makes it easier to understand more complex viral features.
“If there are multiple mechanisms at work, it gets a little trickier,” Johnson said. “My angle is more generic, which makes it easier to tease them apart.”
Lauren and Claire Gibbs share contagious laughter, ambition and a charismatic sarcasm.
Both are honor students at Shawnee Mission East High School in a Kansas City suburb.
They also share a neuromuscular disease called spinal muscular atrophy (SMA), designated as an “orphan disease” because it affects fewer than 200,000 people in the U.S.
However, the landscape for individuals with SMA is quickly changing with the development of new drugs.
More than 7 million people in the United States are carriers (approximately 1 in 40) of the so-called “rare” neurodegenerative disease, SMA.
Lauren,17 (left) and Claire, 16 (right), say their shared SMA diagnosis has strengthened their relationship and presented them with opportunities to travel and share their experiences. | Photo provided by the Gibbs family.
Faces of SMA
The success of therapeutics in lab experiments provides a new layer of hope for individuals and families living with the disease.
Lauren, now 17, fit the criteria for SMA Type III, while Claire, now 16, showed symptoms of a more severe manifestation of the disease, SMA Type II.
Lauren and Claire Gibbs were diagnosed on the same day.
Despite their numerous similarities, the biggest disparity between them is mobility.
Claire uses a power wheel chair while Lauren is able to use a manual chair. It’s not unusual to see Lauren being pulled along in her chair, playfully hanging onto the back of Claire’s motorized chair.
Lauren is participating in a clinical trial with ISIS-SMNRx a compound developed by Isis Pharmaceuticals, a leading company in the antisense drug discovery and development based in Carlsbad, Calif. Lauren feels that she has gained stamina and a greater ability to walk — a feat that wasn’t routine just five years ago.
Prior to the trial, Lauren was able to walk only for short distances.
Tim and Natalie Gibbs with their daughters Lauren, 17 (left) and Claire, 16 (right) in Washington D.C. The Gibbs have been visible advocates in the fight for a cure for spinal muscular atrophy. | Photo provided by the Gibbs family.
Bringing New Hope
A new experimental drug developed by researchers at the Christopher S. Bond Life Sciences Center, is bringing hope to individuals with the orphan disease affecting one in 6,000 people.
Christian Lorson PhD, investigator in the Bond Life Sciences Center and Professor of Veterinary Pathobiology at the University of Missouri, has been researching SMA for seventeen years and has made a recent breakthrough with the development of a novel compound found to be highly efficacious in animal models of disease. In April, a patent was filed for Lorson’s compound for use in SMA.
Lorson’s therapeutic, an antisense oligonucleotide (a fancy name for a small molecule therapeutic that falls under the umbrella of gene therapy), repairs expression from the gene affected by the disease. The research was published May in in the Oxford University Press, Human Molecular Genetics.
The drug developed by Lorson’s lab is conceptually similar to ISIS-SMNRx already in clinical trial developed by Isis Pharmaceuticals and a team of investigators at Cold Spring Harbor Laboratory headed by Dr. Adrian Krainer.
Antisense drugs are not a new practice, but their wide-spread adoption seems to be on the cusp with recent success stories like the commercialization of an FDA-approved antisense compound produced by Isis in 2013 called Kynamro for the treatment of homozygous familial hypercholesterolemia, a high cholesterol disorder that is passed down through families.
Science behind success
The National Institutes of Health has listed SMA as the neurological disease closest to finding a cure. Discoveries made by the Lorson Lab have contributed significantly to current scientific understanding of the disease mechanisms and to the advances being made in finding an effective treatment for SMA.
These antisense therapies work because of the genetic makeup of SMA —the genetics are incredibly clear: a single, specific gene called Survival Motor Neuron 1 (SMN1) has been pinpointed as the cause of SMA.
SMA is a neurodegenerative disorder, meaning muscles become weaker over time due to sick or dying neurons.
These neurons become less functional because of low levels of the SMN.
Remarkably, the disease can be reversed in animal models of disease if the nearly identical duplicate gene, SMN2, can be “turned on” to compensate for low SMN levels.
Lorson’s antisense oligonucleotide therapeutic provides incredible specificity because it hones in on a specific genetic target sequence within SMN2 RNA and allows proper “editing” of the RNA encoding the SMN protein. The strategy is to “repress the repressor,” Lorson said.
The SMA-specific defect lies at the RNA step – the “cutting and splicing” of important RNA sequences does not happen efficiently in SMN2 RNAs because of a several “repressor” signals.
“The final chapter of the book — or the final exon — is omitted,” Lorson said. “But the exciting part is that the important chapter is still there – and can be tricked into being read correctly: if you know how.”
The new, antisense oligonucleotide seems to know how to get the job done.
The existence of such similar genes as SMN1 and SMN2 in humans creates a rare genetic landscape lending itself especially to a therapeutic development for SMA.
Humans are unique in this duplication — something Lorson calls a “genetic happenstance” that, on an evolutionary scale, may as well have happened yesterday.
Why humans have developed this redundant gene is unknown.
Thalia Sass, an MU biological sciences major, genotypes samples in the Lorson Lab where spinal muscular atrophy is researched.
Timing is everything
In addition to the developments of new SMA therapeutics, Lorson and his lab sought to answer an important biological question concerning the disease: When can a therapeutic be administered and still show some degree of efficacy?
Lorson’s research found that the earliest administration of a treatment provided the best outlook— extending the survival of laboratory mice by 500 to 700 percent, “a profound rescue,” according to his research published in April in the Oxford University Press, Human Molecular Genetics.
A near complete, 90 percent rescue was demonstrated in severe SMA mouse models. But even when the therapeutic was administered after the onset of SMA symptoms, there was still a significant impact on the severity of the disease.
“If you replace SMN early and get (a therapeutic) to cells that are important to the disease, you correct it,” Lorson said. “This provides hope that patients who have been diagnosed will still see some therapeutic benefit even if it is clear that the best results will likely come from early therapeutic administration.”
In Lorson’s study it’s definitive that the earlier a therapeutic can be administered, the better the outcome for individuals with SMA.
“This really points towards a strong push for neonatal screening,” Lorson said. “Infant screening would likely be incredibly beneficial for SMA and that’s something that the SMA community is really excited about.”
A breakthrough for families
On June 2, Lauren and Claire Gibbs attended a routine, annual rehab appointment with Dr. Robert Rinaldi, MD, division of pediatric rehabilitation medicine and attending physician at Children’s Mercy Hospital in Kansas City, Mo Dr. Rinaldi is not associated with the Isis clinical trial.
The appointment was like a reunion among close friends — Rinaldi began seeing Claire and Lauren Gibbs 16 years ago, the first year that he began working at the hospital and when the girls were one- and two-years-old, respectively.
The girls did all of the routine tests —measuring strength of grip and breathing, and assessing range of movement with the occupational and physical therapists.
A little later, Rinaldi sat with Natalie Gibbs, Lauren and Claire’s mother and a relentless advocate for advancement in SMA awareness.
Typically the muscles of individuals with SMA deteriorate over time, but together they inspected the definition of a new calf muscle on Lauren’s left leg.
For a young woman with Type III SMA — this means she can walk for short distances with little discomfort but still uses her wheel chair a majority of the time — Lauren’s new calf muscle is a remarkable achievement.
As Lauren continues to participate in the ISIS antisense therapy clinical trial, her conditions continue to improve dramatically, even with the late administration of the therapy — in her case, 16 years after her diagnosis and onset of effects.
Lauren believes her ability and stamina for walking have increased significantly.
“Quite frankly my jaw almost hit the ground when she stood up — the change was that impressive to me,” Rinaldi said.
Rinaldi, also the co-director of the Nerve and Muscle Clinics at the hospital, had last seen Lauren two years ago. He said the Lauren he saw during a routine rehab appointment in June was like seeing a new person altogether.
“The way she stood up from the wheel chair — how quickly she did that with no support — her posture when she was standing up was more upright, her pelvis was in a much better position, her core was straighter,” Rinaldi said. “It struck me immediately how much better she looked.”
Lauren Gibbs is the first of Rinaldi’s patients to have participated in the ISIS clinical trial.
“It’s moving very fast in this field,” Rinaldi said. “I think the technology that’s evolving in research is opening up more avenues for investigation for us and there’s a big desire to find a cure for these types of diseases.”
The progress has rewarded the Gibbs family’s advocacy in SMA awareness and they’ve been able to set new goals they didn’t imagine were possible when the diagnoses for Lauren and Claire were made. Natalie Gibbs is a long-time member of Families of SMA and is currently on their Board of Directors.
The organization Families of SMA is currently providing funding to Lorson to advance this research area.
“We’re able to see first hand — and our physician who has been watching them for sixteen years has seen — that everything we’re doing in the clinical trials is really making a difference,” Natalie Gibbs said.
Over the course of their daughters’ lives, Natalie and her husband Tim Gibbs say a shift in momentum has accelerated the technology and research toward finding a cure for SMA.
“I am really impressed with the progress Lauren has made with the trial and how well Claire is doing overall,” Natalie Gibbs said. “Even though it’s a progressive and very devastating type of disease, I feel like we’re really conquering it.”