By Caleb O’Brien | MU Bond Life Sciences Center
Jean Camden and Luke Woods have an ant’s-eye view of Alzheimer’s disease.
Both are bench scientists in the laboratory of Gary Weisman, a professor of biochemistry at the Bond Life Sciences Center. Jean has spent the past 12 of her 35 years at the University of Missouri in the Weisman lab, running experiments, managing the lab and working with students. Luke joined the Weisman lab six years ago, doing what he call’s the dirty work of science: “Gary does the writing and the NIH stuff, but down in the trenches — that’s me and Jean.”
Weisman’s lab studies Alzheimer’s and other diseases, so I sat down recently with Jean and Luke to talk about their research for Alzheimer’s & Brain Awareness Month.
Q: What does your lab do, and how does it involve Alzheimer’s?
Luke: We primarily have two projects. One, which has been a very longstanding project, is focused on salivary glands and salivary gland inflammation. The other is the Alzheimer’s project. The link between them is a particular type of cell surface receptor called a nucleotide receptor — more specifically, a P2 nucleotide receptor called P2Y2. These P2 receptors function in a lot of different ways, but the link is with inflammation: We look at P2 receptors in salivary gland inflammation and in Alzheimer’s disease, which has a very large inflammation component that often gets glossed over. In a lot of Alzheimer’s articles that the public reads, you hear about amyloid beta plaques and tau tangles and neurodegeneration, but a large component of that is inflammation, where some of the resident non-neurons in the brain start responding like there’s inflammation in the brain, and it actually kills neurons. That’s been the focus in Gary’s lab for the past 30-plus years.
JEAN: The P2 receptors — especially the P2X7 and P2Y2 which we focus on — Gary during his postdoctoral work started studying these receptors without really knowing that they existed. At the time, he just knew that there was a pore formed in cells caused by the addition of the nucleotide ATP which eventually leads to apoptosis (cell death). Eventually, we cloned the human P2Y2 receptor gene with another group in North Carolina, so we call it “our receptor.” It only appears in cells under inflammatory conditions, such as Alzheimer’s disease, salivary gland autoimmune disease and cardiovascular disease. Any time you have tissue damage, it looks like the P2Y2 receptor is up-regulated. And then once the damage is healed, the receptor goes away.
Inflammation is good — we want inflammation, that’s how we heal — it’s the chronic inflammation that’s bad. But we really don’t know how these receptors work and what their role is during chronic inflammation. Do we want to activate them, or do we want to inhibit them?
LUKE: Scientists have investigated P2X7 receptor antagonists in the treatment of Crohn’s disease and rheumatoid arthritis — there are several clinical trials that have been focused on these receptors, evaluating whether you want to block or activate them. If you block them, you prevent the acute inflammatory responses that are good for wound healing; if you activate them, you may extend those responses past the healing phase into a chronic inflammatory phase that can be quite damaging. So unraveling that fine line of what you want to be doing to these receptors in disease settings is sort of what we do here.

Q: When I think of Alzheimer’s, I think of a shriveled, shrunken brain, but I associate inflammation with swelling. Why the difference?
LUKE: I think the distinction is acute versus chronic inflammation. With acute inflammation, you get swelling. The body has different types of immune responses: acute responders like neutrophils and macrophages are immune cells that act quickly. They come in, for example, if you have a scratch, and there can be swelling. Along with macrophages neutrophils can protect cells from bacteria. The macrophages can also clean up damaged tissue and then the repair cells go to work. Cells come in that lay down a new matrix, whereas undamaged cells then migrate onto the matrix and regenerate. Well, what happens after you’re done repairing is that there are signals that tell the inflammation to stop. In chronic inflammation, that’s where you have continued cell death, and the tissue would then shrivel up. The shriveled brain that you’re referring to is during chronic inflammation, and that’s an end-of-life case, after a very long bout with Alzheimer’s.
JEAN: What we think of as inflammation is often a cut or a wound. It’s only been in recent years that Alzheimer’s disease has been considered an inflammatory disease. We have a phenomenal immune system, but when it goes awry, you have problems. In the other disease we look at — an autoimmune disease — your immune system starts to attack your own body. It’s hard to treat and understand the underlying mechanism.
Q: So how are you trying to unravel the role of inflammation in Alzheimer’s?
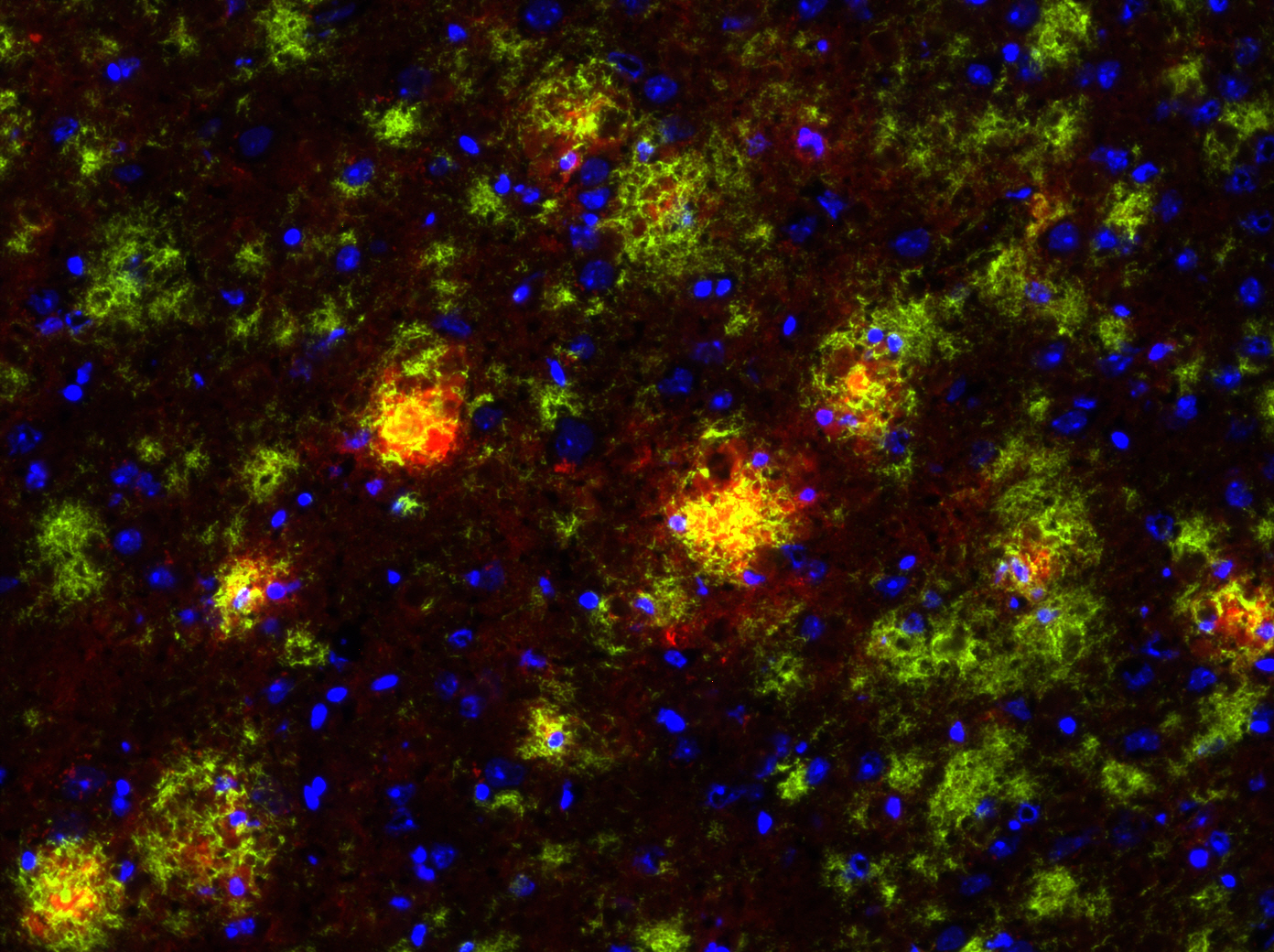
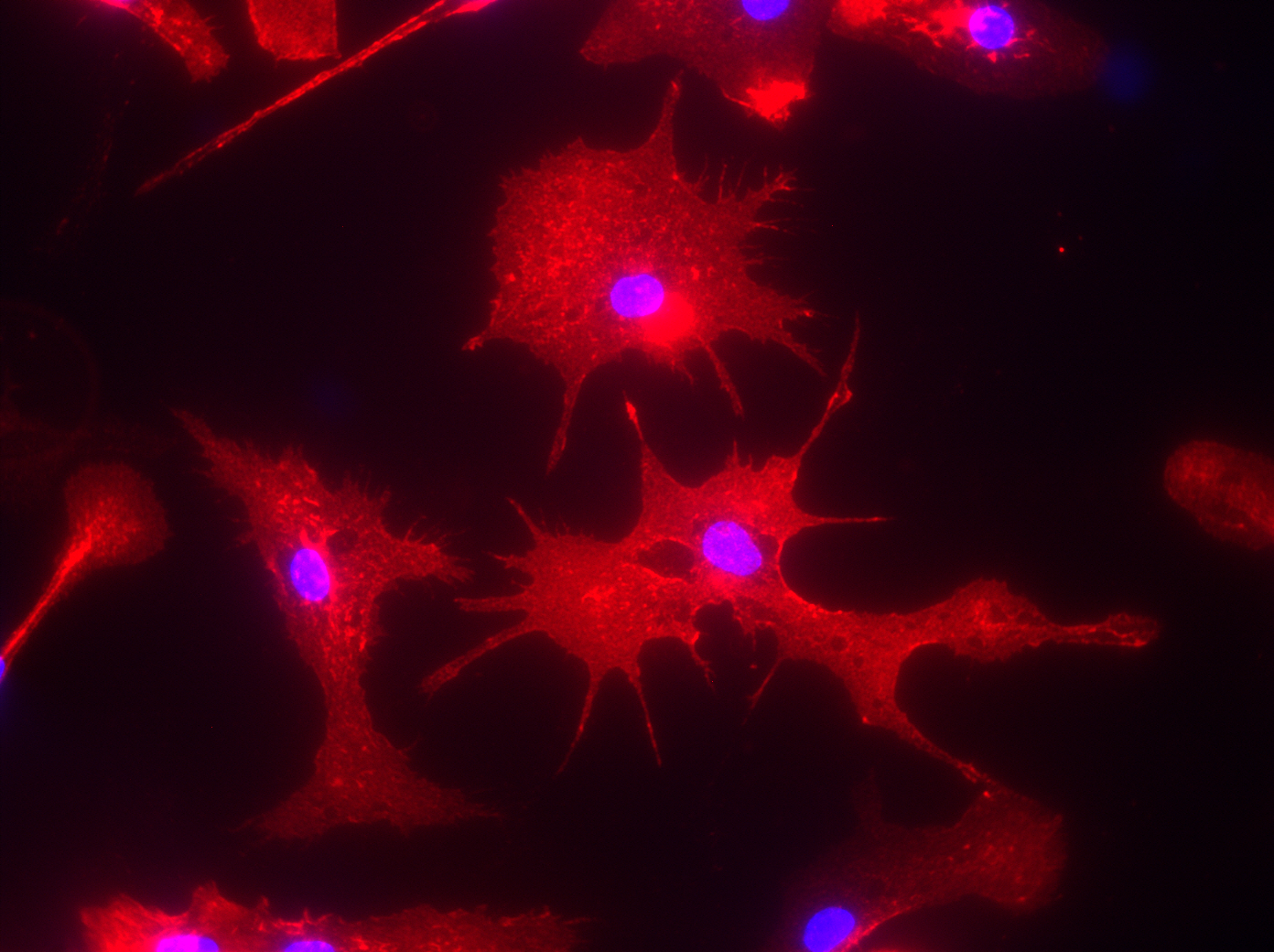
JEAN: To study Alzheimer’s, we have an Alzheimer’s mouse model. It overexpresses a gene for the amyloid precursor protein that enables the brain to accumulate high amounts of beta-amyloid plaques that you always hear about. So we’re using this mouse model that we’ve crossed with a mouse that does not express any P2Y2 receptor, so it’s called a knockout mouse. The P2Y2 receptor knockout mouse by itself is fine, and the Alzheimer’s mouse does develop Ab plaques, but it lives to approximately 6 months old before it will develop behavioral defects. The interesting thing is that when we cross the P2Y2 receptor knockout mouse with the Alzheimer’s mouse, the offspring that are Alzheimer’s mice without P2Y2 receptors prematurely die. So at least in this Alzheimer’s mouse model, it looks like the presence of the P2Y2 receptor is protective, because without it, the Alzheimer’s mice die much earlier. But we don’t really know which cell type is most important: Is it the P2Y2 receptor up-regulated on neurons that acts to repair them —which we’ve already shown happens — or is it the P2Y2 receptor on microglia (an immune cell of the brain), or is it the P2Y2 receptor on blood vessels in the brain that help recruit immune cells from the cardiovascular system to help with repair?
So we’re using this mouse model to investigate the role of the P2Y2 receptor, plus we also use cell lines because we can easily control the environment for these cell lines in culture. We isolate primary neurons, we can prepare primary microglial cells or we can purchase cell lines that comprise blood vessels. We can then utilize these tools to investigate cell signaling mechanisms for the P2Y2 receptor in individual cell types.
LUKE: One of the findings that we have found interesting in these primary cells is when you take them fresh out of the mouse, put them in a dish and then treat them as you wish. We’ve shown that if you activate the P2Y2 receptor in primary microglia from the mouse, they will actually engulf and chew up beta-amyloid. And so one of the things we think might be happening in this Alzheimer’s mouse model is that P2Y2 receptor activation in these microglial immune cells in the brain is working to break down those beta-amyloid plaques. And when you lose the P2Y2 receptor in that mouse model, those plaques develop quicker because the immune cells are no longer offering protection by chewing up that beta-amyloid. That’s one of the hypotheses we’re exploring right now.
Q: So you’d bet that these receptors are actually protective against Alzheimer’s?
JEAN: Yes. Going back to the human — it’s hard to get human tissues, especially brain tissues, but there is one published study that has shown that in Alzheimer’s patients who have passed away the P2Y2 receptor is down-regulated, meaning there’s not much left. Which would make sense. If it’s down-regulated, the plaques aren’t able to be chewed up, per se, by these microglia. There’s a correlation between low levels of P2Y2 receptors and Alzheimer’s disease that is apparent at the end of life.
LUKE: It’s very difficult to do some of these studies in humans because most of the available Alzheimer’s tissues are from end of life cases where you can only look at the end result of the disease without looking at the progression of the disease. Obviously you can’t take brain tissue from a living person, so the ability to study live cells from Alzheimer’s patients is limited. We rely very heavily on mouse models.
Q: What have been the biggest shifts in our understanding of Alzheimer’s in recent years?
LUKE: Maybe one shift — I may not be the best expert to speak about it — is the idea that the beta-amyloid plaques are the cause of disease. It is now being mostly recognized that they’re really the tombstones of the disease. They’re not the initial cause, but rather the end result of the disease. For a long time investigators were focused on trying to prevent the buildup of beta-amyloid because that was one aspect of Alzheimer’s disease that you could see and measure. Now the thinking is that maybe the beta-amyloid does not contribute as much to disease progression as originally thought, and rather is the end result of a complicated mechanism that is actually causing the neurodegeneration.
JEAN: There’s still debate on what causes Alzheimer’s disease. There is a small percentage of patients where it’s actually related to a genetic alteration in the amyloid precursor protein gene.
LUKE: Another link has been with the ApoE (apolipoprotein E) gene, which makes a lipoprotein and cholesterol transporter. We inherit 1 copy of the ApoE gene from each of our parents and it has been shown that individuals who have at least 1 copy of a particular variant of the gene called ApoE4 are at increased risk of developing Alzheimer’s disease.
Q: From the perspective of a lab scientist, why do you care about Alzheimer’s?
JEAN: We care about any disease, really, and if we can show that our receptors have anything to do with any disease, we’d be proud to have a role in that.
LUKE: We don’t do much clinical science here, it’s mostly basic science. We contribute to the basic understanding of the disease so that drug companies and medicinal chemists who develop drugs for clinical use in Alzheimer’s patients can say, “Hey, this group’s research found a new mechanism related to Alzheimer’s disease, so let’s target this pathway to treat the disease.” It’s always nice to contribute to that sort of ground-level science.
JEAN: That would be the ideal, to show that whether you have to activate or inhibit the P2Y2 receptor, it does something to improve the clinical outcome in Alzheimer’s patients. A better understanding of Alzheimer’s and other diseases is what’s needed — we’re just working to provide a piece of the puzzle.
Q: How has being down in the trenches changed your perspective on research and Alzheimer’s in general?
JEAN: We’re the ones who are hoping to clarify the direction for science to go. We do the experiments and we are the first ones to see the data. We collect the data that becomes the cornerstone for deciding the direction our research goes. I think Gary would agree with that — he depends on us a lot to collect the data and we depend on him to help determine which scientific findings to chase and which ones not to chase.
I’ve been doing this for 35 years, and I really do enjoy the science. I’ve seen the science of these nucleotide receptors come a long way. These receptors have in common their use of extracellular nucleotides, particularly ATP (or adenosine triphosphate, more commonly known as the intracellular high energy molecule of all cells). And this ATP, is at a high concentration inside cells, so when it is released by cell damage, it can easily activate nucleotide receptors on nearby cells. It was Dr. Geoffrey Burnstock, now considered to be the grandfather of nucleotide receptors, who claimed a long time ago that there are receptors on the outside of cells that respond to ATP. Everybody kind of laughed at him, “Yeah, sure, right. There’s no way: ATP belongs inside the cell.” So for me personally, to come in on the ground level for these receptors and find a role for them in a variety of diseases has been exciting for me.
LUKE: ATP is the energy currency inside of all cells, so it’s use outside cells would be like tossing money out the window. Why would they want ATP outside the cell? It didn’t make any sense at the time, but looking back I think it does. What happens if you damage or rupture a bunch of cells during an injury? You get the release of a high concentration of ATP that neighboring cells recognize as a danger signal telling them that an injury has occurred. In that sense, ATP makes the perfect signaling molecule to tell other cells that an injury has occurred and they need to start the repair work by recruiting immune cells to the damaged tissue.

Q: Where would you like to be in five years with this research?
JEAN: I talked about determining how the P2Y2 receptor in this mouse model was protective. It would be nice to find out which cell type on which the P2Y2 receptor is expressed in contributes most to neuroprotection. Our hypothesis would be that the microglial cells are very important, since they gobble up beta-amyloid, but other cell types including neurons and endothelial cells are likely involved. We’re also anxious to look at other inflammatory diseases to see if the P2Y2 receptor plays a similar role there.
LUKE: From somebody who does a lot of bench work, something I would like to see is a really good tool, a specific agonist or antagonist of the P2Y2 receptor that could be used in the clinic. There are a few suitable compounds available that we use to investigate the P2X7 receptor— I’ve told you that some have been tested in clinical trials — but the P2Y2 receptor has been sort of an enigma, due to the lack of selective inhibitors and agonists that are specific enough for clinical use. I’d like to see the development of a specific agonist or antagonist that could eventually be used to treat inflammatory diseases. There’s no reliable drug that is currently suitable to investigate the P2Y2 receptor in animals or humans, so clearly more work is needed there.
This interview has been edited for length and clarity.