Michael Arowolo is a visiting professor in the lab of Dong Xu, a Bond LSC principal investigator. | photo by Braiden Wade, Bond LSC
The African proverb “it takes a village to raise a child” can especially apply in science where that village includes mentors like Dong Xu, a Bond Life Sciences Center principal investigator, who has trained hundreds of students and collaborators.
Michael Arowolo, is among those mentored, having spent the past two years in Xu’s lab as a visiting scholar. In August, he will take that experience with him to Xavier University of Louisiana as a new assistant professor.
“Dr. Xu has really impacted me in so many facets of my life,” said Arowolo. “He has given me the privilege of tapping into his knowledge.”
The Nigerian native landed at Mizzou because around 40 scientific publications credited to him caught Xu’s attention, and he was invited to be a visiting professor in July 2022. He quickly stood out amongst other people Xu had worked with.
“In Africa, the research infrastructure is not as advanced as the U.S. In that environment, if he could publish that many papers, I was very impressed,” said Xu, also head of Mizzou’s Digital Biology lab. “He’s a self-starter, he actually takes initiative. I don’t need to motivate him to work hard.”
With experience from earning his Ph.D. in Nigeria, Arowolo scrambled between roles as a lecturer, researcher, exam officer and more. Working at the Bond LSC was a change of environment for him that he was excited to take on. He has made it a practice to come into the lab around 7:30 a.m. and leaves around 5 p.m. to meet his work needs. He spends his time mostly working on his computer, coding.
While visiting professors typically stay less than a year, Arowolo’s stay was extended because felt he still had more to contribute.
Arowolo’s research focus at Mizzou has been discovering innovations in biological pathways. To break it down, he collects information on gene interactions into a database using artificial intelligence to develop models such as Siamese Neural Network for identification of relevant genes. That information can help scientists access a multitude of information in one place to create targeted treatments and drugs for diseases.
“They won’t need to go through the back end and stress themselves with ‘What is all this computational jargon, what is all this coding?’” he said. “They can have a platform that they can easily interact with…and get the results they need to get.”
AI has become a major part of Arowolo’s work. He is developing his own large language model, using an advanced retrieval augmented generative mode that identifies and recognizes pertinent genes and describes its relationships with specific biological processes in human cells.
He recognizes how AI has been taking the world by storm, and he wants to use it to help people.
“Instead of just thinking that the world is over, that AI will take over, before AI takes over, we will tap into AI and be the speaker for AI,” he said.
Arowolo has also expanded past his computational work and has been collaborating with the Mizzou School of Medicine on a new project. His team proposed a medication dispensing machine that would help Alzheimer’s patients. His team is currently in the process of developing a product sellable to big companies like Amazon, he said.
“He transformed academic work into a commercial product,” Xu said.
But, on top of his research, Arowolo teaches undergraduate and graduate students and mentors Ph.D. students.
“It’s become a passion,” he said. “I’ve mentored over 100 students, and they are also doing well in their area of endeavor, most of them in Dr. Xu’s lab.”
Now, Arowolo will pass down what he’s learned from Dr. Xu to his new students at Xavier University of Louisiana in August.
Xu, on the other hand, is excited to see Arowolo take on this next step.
“I think the main reason you train people is not only so they will create a product but we hope to help them move onto a more independent position with a higher salary,” Xu said.
When Arowolo reflects on what he has accomplished so far, he envisions the work of him and his colleagues as the tool that will help people get access to the medications they need.
“I know there will be a day that will come, and we’ll have the right solutions to consider these diseases,” he said.
Investigators at Bond LSC take steps to apply basic research
By Cara Penquite | Bond LSC
Photo by Lauren Hines | Bond LSC
Scribbling in a lab notebook and planning experiments tucked between shelves of equipment, it’s easy to fixate on day-to-day lab operations. But scientists also face the challenge of finding how research can improve the world around us.
“The direction, the vision of the lab, ultimately comes from the principal investigator that bridges the research into applied directions,” said Jay Thelen, biochemistry professor and Bond LSC principal investigator
Despite the focus on basic research within the Bond LSC, many principal investigators choose to take their research to the next level with commercial partnerships.
Thelen’s lab researches ways to increase oil production in seeds and has three patents licensed to Yield 10 Bioscience, a sustainable crop innovation company who applies Thelen’s research to commercial crops. While seed oils like canola and soybean oil are known for their use in cooking, Thelen explains that increased production of these oils could play a larger role in sustainable fuel sources such as biodiesel and sustainable aviation fuel.
“We have to make more oil to balance out our need to eat it [and] our need to wean ourselves off of fossil fuels,” Thelen said. “To do that, we need to either plant more acres of oil seeds, or we have to raise the oil in existing oil seeds.”
Thelen researches enzymes with that potential application in mind. One is acetyl-CoA carboxylase, the enzyme which initializes the production of fatty acid chains found in plant oils.
“We’ve known this is an important enzyme, and we know that any tinkering you do with it has an impact on the oil production,” Thelen said. “In this case we’ve made new discoveries that permitted us to rationally engineer this enzyme to make it more active.”
Thelen suggests thinking of the enzyme as a “gatekeeper” to oil production which initializes the production of fatty acids and increases oil production. Thelen’s lab identified two different gene families that influence the activity of the enzyme in Arabidopsis and camelina plants. Yield 10 then applies these discoveries in other commercial plants.
While Thelen works closely with his commercial partner — having served on their scientific advisory board for three years and now stays in contact with Yield 10’s CEO to develop research projects — some labs stick with short-term arrangements.
Kamlendra Singh — assistant director of the Molecular Interactions Core at Bond LSC and Veterinary Pathobiology research assistant professor — studies HIV treatments. His lab identified a compound licensed by a commercial partner that targets the shell containing the virus’ genetic information.
Singh’s work in HIV started in 1994 with basic research investigating the enzyme that makes the viral DNA.
“I wasn’t into [studying] the drugs when I started working on HIV, I was mostly trying to understand how HIV enzymes works,” Singh said. “Once you know how the enzyme works, then you can target these enzymes for discovering the drugs.”
After years of studying how the enzyme works, Singh switched to HIV treatment. The first step to develop a treatment is to look for structures in the virus that the drug could potentially target to stop the viral replication. Singh targeted the shell around the virus’ genetic information known as the HIV capsid.
Building on previous research, Singh’s lab developed a compound able to bind the HIV capsid and prevent it from releasing the contained genetic information. Even with the licensing of his compound, Singh plans to continue researching ways to improve it.
“There are two reasons to keep working on it. One, well it’s my brainchild,” Singh said. “The second reason is as the company grows, we grow. We get more recognition and more funding. You can use it to [study] different viruses or use the same funding to improve upon it.”
While Singh plans to remain looking towards the applied side of his HIV research, he does not forget his roots in basic research.
“You have to put in time … [to] understand the system first, which is basic science, before you go to applied science,” Singh said.
Michael Roberts, a Chancellor’s Professor Emeritus of animal sciences and biochemistry who has had several patented projects, focuses on improving basic science projects and applies for patents if warranted.
“I don’t deliberately go into anything for commercial purposes,” Roberts said. “If I see something that I think does have commercial application, I’m happy to do it, but that is usually after you do [basic sciences].”
Whether starting a project with applications in mind or focusing on basic research, knowledge gained through research can be building blocks for the future.
“Science is simple. Even the most applied research project has its genesis in basic biology and basic research,” Thelen said.
DNA is the genetic material that determines the characteristics of plants and animals. Using CRISPR gene editing, researchers altered the characteristics of rice plants. | Creative Commons Photo by Pixabay
By Cara Penquite | Bond LSC
A tickle in the throat, a stuffy nose, congestion . . . the tell-tale signs of a cold are familiar to most, and many know that with enough rest, the immune cells on standby in the human body will destroy any invaders. But what happens when plants get sick?
The Bing Yang Lab from the Bond Life Sciences Center at the University of Missouri studied this question with the Reuben Peters lab at Iowa State University by analyzing the purpose of gene clusters in rice. Along with the Zhaohu Li lab at China Agricultural University, the researchers — utilized CRISPR gene editing to determine the function of certain gene clusters in rice.
“I thought this could be something very important,” Yang said, “We can take out a huge amount of DNA to see if the gene cluster could play a role in this resistance [to disease].”
CRISPR – a gene editing tool – allows the researchers to change the DNA of the rice plants by removing sections of genetic code, known as gene clusters. By removing the cluster, scientists can see what breaks in the plant, thus identifying each cluster’s role in the plant’s health.
“These were what we termed biosynthetic gene clusters. They have unrelated genes that are close together, their physical proximity on a chromosome, and they all act in one single pathway to make a small family of natural products,” Peters said.
The scientists compared unedited rice plants with rice plants missing the gene clusters, and they determined that those without the gene clusters had decreased resistance to certain diseases. This was due to the decreased production of antimicrobial — or pathogen fighting — natural products.
The genes code for enzymes that create labdane-related diterpenoids — natural products responsible for protecting plants from infection, termed phytoalexins — a defense response.
The loss of these compounds made the plants less resistant to disease. While humans have mobile cells that can travel to pathogens through the immune system, plants rely on local chemical responses to fight off invaders.
“They’ll recognize components of the fungal cell wall, or they’ll interact with specific proteins from the fungus or the bacteria, [and] that triggers what we consider to be an immune response,” Peters said. “They have to rapidly make these compounds in what would otherwise be a general photosynthetic cell or just an epidermal cell on the surface of the leaf.”
By identifying which genes produce certain chemicals, the researchers have a greater understanding of the plant immune response, and they can manipulate the genetics of the plants to create stronger immune responses.
“We can fine tune those genes, which could increase the [protections] and make the plant more resistant to the pathogen,” Yang said.
Peters suggested since the scientists know which genes code for these proteins, they can make the proteins and create an antibiotic to prevent infections in crops and livestock.
“We do think that some of these compounds are going to be important for potential pharmaceutical activity,” Peters said. “Not necessarily the ones in rice, but related compounds, and that’s another reason we studied this.”
This research published online in New Phytologist October 16, 2021, under the title “Dissecting the labdane-related diterpenoid biosynthetic gene clusters in rice reveals directional cross-cluster phytotoxicity.”
One class changed senior K’Imani Davis’s mind, who is now going into her senior year working in the Anand Chandrasekhar lab at Bond LSC.
“I used to actually hate science, and when I say hate, I hated it,” Davis said. “Senior year of high school I took AP Bio, I loved it. I don’t know what happened, but I started to try and I liked the subject.”
After Davis’s change of heart, she decided to start out at MU as a biology major, and she is now going the pre-med route.
Just like Davis ended up in science, she ended up at MU by chance.
“I wasn’t going to come here at first,” Davis said. “I never visited and never knew someone here. I was so indecisive about my college decisions so I put all my options in a hat and just drew, I drew Mizzou so I just came here.”
Davis came to MU knowing nothing about the campus, programs or any students. And then she stumbled across an opportunity that changed her college experience for her.
“I actually got involved by accident,” Davis said. “I had a class in this building and across from my class was the undergraduate research office. I decided I wanted to do research because it was science out of the classroom.”
From freshman year to now, Davis has found a home away from home in her lab.
“I think of my lab as a family,” Davis said. “We always have something to do all together every semester. It’s very family oriented, it’s very close and tight knit. My boss sometimes even acts like my father.”
Davis didn’t just find a family at Bond LSC, she found her passion. The lab is studying neuronal migration in zebra fish. This study looks at how neurons move into place as the brain and nervous system develops with the hope of learning how the process works and using that information to understand neuro-degenerative disease. In her freshman year when Davis was an intern in the lab, she fed the fish and cleaned the tanks. Now, she analyzes fish behaviors and compares swim patterns between wild and mutated zebra fish.
“By analyzing their behavior, we are able to see if there are neuronal migration patterns, and we see what defects leads to different behaviors,” Davis said.
Davis’s goal for this upcoming year is to have a poster of her research to display to others. Her experience at MU has brought her a family, a passion and no longer a hatred for science.
“I think everything happens for a reason,” Davis said.
Scientists explore genetic similarities between plants and mice
University of Missouri PhD Candidate Daniel L. Leuchtman peers through an Arabidopsis plant. Leuchtman has been experimenting with replacing a gene in the plants immune system with a similar gene from mice. | Photograph by Justin L. Stewart/MU Bond Life Sciences Center
By Justin L. Stewart | MU Bond Life Sciences Center
Almost two-thirds of what makes a human a human and a fly a fly are the same, according to the NIH genome research institute.
If recent research at the University of Missouri’s Bond Life Sciences Center is verified, we’ll soon see that plants and mice aren’t all that different, either.
Dan Leuchtman studies a gene in Arabidopsis plants called SRFR1, or “Surfer One.” SRFR1 regulates plant immune systems and tell them when they are infected with diseases or illnesses. Leuchtman studies this model plant as a Ph.D. candidate at MU, splitting time between the labs of Walter Gassmann and Mannie Liscum.
His research involves breeding Arabidopsis plants missing the SRFR1 gene and then replacing it with the MmSRFR1 gene.
A series of Arabidopsis plants show the differences between the plants, from left, without SRFR1, with MmSRFR1 and with SRFR1. | Photograph by Justin L. Stewart/MU Bond Life Sciences Center
So, what is MmSRFR1? Leuchtman and company believe it’s the animal equivalent of SRFR1, though they aren’t fully aware of all of its’ functions.
“We’re actually one of the first groups to characterize it,” Leuchtman said.
Arabidopsis plants missing the SRFR1 gene struggle to grow at all, appearing vastly different from normal plants. Leuchtman says that a plant missing the SRFR1 gene is a mangled little ball of leaves curled in on itself. “It’s really strange looking.”
While his experiments haven’t created statuesque plants equal to those with natural SRFR1 genes present, the Arabidopsis plants with MmSRFR1 show a notable difference from those completely lacking SRFR1. Leuchtman says the plants with MmSRFR1 lie somewhere in between a normal plant and one lacking SRFR1.
University of Missouri PhD Candidate Daniel L. Leuchtman poses for a portrait in a Bond Life Sciences Center greenhouse. Leuchtman has been experimenting with replacing a gene in Arabidopsis plants immune system with a similar gene from mice. | Photograph by Justin L. Stewart/MU Bond Life Sciences Center
“At its’ core, it’s more understanding fundamental biology. How do we work? How do organisms tick? How do you go from DNA in a little bag of salts to a walking, talking organism?” Leuchtman said. “The more you know about how an organism functions, the more opportunities you have to find something that makes an impact.”
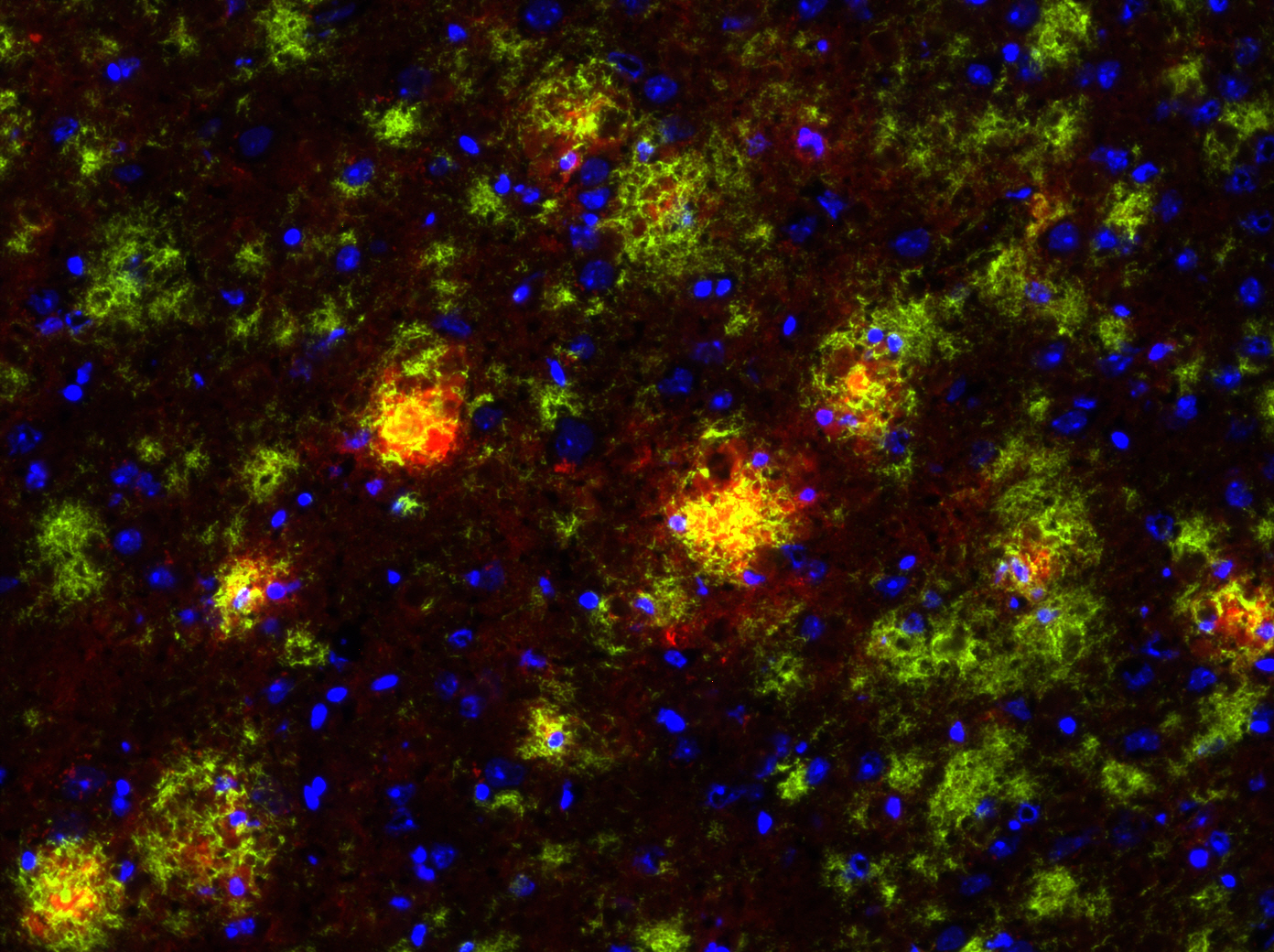
This immunofluorescence picture shows the brain of an Alzheimer’s disease mouse model, also known as the TgCRND8 mouse. In the picture, the amyloid beta plaques are stained green and the microglia, or immune cells of the brain, are stained red. Image courtesy of Luke Woods.
By Caleb O’Brien | MU Bond Life Sciences Center
Jean Camden and Luke Woods have an ant’s-eye view of Alzheimer’s disease.
Both are bench scientists in the laboratory of Gary Weisman, a professor of biochemistry at the Bond Life Sciences Center. Jean has spent the past 12 of her 35 years at the University of Missouri in the Weisman lab, running experiments, managing the lab and working with students. Luke joined the Weisman lab six years ago, doing what he call’s the dirty work of science: “Gary does the writing and the NIH stuff, but down in the trenches — that’s me and Jean.”
Weisman’s lab studies Alzheimer’s and other diseases, so I sat down recently with Jean and Luke to talk about their research for Alzheimer’s & Brain Awareness Month.
Q: What does your lab do, and how does it involve Alzheimer’s?
Luke: We primarily have two projects. One, which has been a very longstanding project, is focused on salivary glands and salivary gland inflammation. The other is the Alzheimer’s project. The link between them is a particular type of cell surface receptor called a nucleotide receptor — more specifically, a P2 nucleotide receptor called P2Y2. These P2 receptors function in a lot of different ways, but the link is with inflammation: We look at P2 receptors in salivary gland inflammation and in Alzheimer’s disease, which has a very large inflammation component that often gets glossed over. In a lot of Alzheimer’s articles that the public reads, you hear about amyloid beta plaques and tau tangles and neurodegeneration, but a large component of that is inflammation, where some of the resident non-neurons in the brain start responding like there’s inflammation in the brain, and it actually kills neurons. That’s been the focus in Gary’s lab for the past 30-plus years.
JEAN: The P2 receptors — especially the P2X7 and P2Y2 which we focus on — Gary during his postdoctoral work started studying these receptors without really knowing that they existed. At the time, he just knew that there was a pore formed in cells caused by the addition of the nucleotide ATP which eventually leads to apoptosis (cell death). Eventually, we cloned the human P2Y2 receptor gene with another group in North Carolina, so we call it “our receptor.” It only appears in cells under inflammatory conditions, such as Alzheimer’s disease, salivary gland autoimmune disease and cardiovascular disease. Any time you have tissue damage, it looks like the P2Y2 receptor is up-regulated. And then once the damage is healed, the receptor goes away.
Inflammation is good — we want inflammation, that’s how we heal — it’s the chronic inflammation that’s bad. But we really don’t know how these receptors work and what their role is during chronic inflammation. Do we want to activate them, or do we want to inhibit them?
LUKE: Scientists have investigated P2X7 receptor antagonists in the treatment of Crohn’s disease and rheumatoid arthritis — there are several clinical trials that have been focused on these receptors, evaluating whether you want to block or activate them. If you block them, you prevent the acute inflammatory responses that are good for wound healing; if you activate them, you may extend those responses past the healing phase into a chronic inflammatory phase that can be quite damaging. So unraveling that fine line of what you want to be doing to these receptors in disease settings is sort of what we do here.
Jean Camden and Luke Woods look at images of a mouse brain with Alzheimer’s disease. // Photo by CALEB O’BRIEN/Bond LSC
Q: When I think of Alzheimer’s, I think of a shriveled, shrunken brain, but I associate inflammation with swelling. Why the difference?
LUKE: I think the distinction is acute versus chronic inflammation. With acute inflammation, you get swelling. The body has different types of immune responses: acute responders like neutrophils and macrophages are immune cells that act quickly. They come in, for example, if you have a scratch, and there can be swelling. Along with macrophages neutrophils can protect cells from bacteria. The macrophages can also clean up damaged tissue and then the repair cells go to work. Cells come in that lay down a new matrix, whereas undamaged cells then migrate onto the matrix and regenerate. Well, what happens after you’re done repairing is that there are signals that tell the inflammation to stop. In chronic inflammation, that’s where you have continued cell death, and the tissue would then shrivel up. The shriveled brain that you’re referring to is during chronic inflammation, and that’s an end-of-life case, after a very long bout with Alzheimer’s.
JEAN: What we think of as inflammation is often a cut or a wound. It’s only been in recent years that Alzheimer’s disease has been considered an inflammatory disease. We have a phenomenal immune system, but when it goes awry, you have problems. In the other disease we look at — an autoimmune disease — your immune system starts to attack your own body. It’s hard to treat and understand the underlying mechanism.
Q: So how are you trying to unravel the role of inflammation in Alzheimer’s?
JEAN: To study Alzheimer’s, we have an Alzheimer’s mouse model. It overexpresses a gene for the amyloid precursor protein that enables the brain to accumulate high amounts of beta-amyloid plaques that you always hear about. So we’re using this mouse model that we’ve crossed with a mouse that does not express any P2Y2 receptor, so it’s called a knockout mouse. The P2Y2 receptor knockout mouse by itself is fine, and the Alzheimer’s mouse does develop Ab plaques, but it lives to approximately 6 months old before it will develop behavioral defects. The interesting thing is that when we cross the P2Y2 receptor knockout mouse with the Alzheimer’s mouse, the offspring that are Alzheimer’s mice without P2Y2 receptors prematurely die. So at least in this Alzheimer’s mouse model, it looks like the presence of the P2Y2 receptor is protective, because without it, the Alzheimer’s mice die much earlier. But we don’t really know which cell type is most important: Is it the P2Y2 receptor up-regulated on neurons that acts to repair them —which we’ve already shown happens — or is it the P2Y2 receptor on microglia (an immune cell of the brain), or is it the P2Y2 receptor on blood vessels in the brain that help recruit immune cells from the cardiovascular system to help with repair?
So we’re using this mouse model to investigate the role of the P2Y2 receptor, plus we also use cell lines because we can easily control the environment for these cell lines in culture. We isolate primary neurons, we can prepare primary microglial cells or we can purchase cell lines that comprise blood vessels. We can then utilize these tools to investigate cell signaling mechanisms for the P2Y2 receptor in individual cell types.
LUKE: One of the findings that we have found interesting in these primary cells is when you take them fresh out of the mouse, put them in a dish and then treat them as you wish. We’ve shown that if you activate the P2Y2 receptor in primary microglia from the mouse, they will actually engulf and chew up beta-amyloid. And so one of the things we think might be happening in this Alzheimer’s mouse model is that P2Y2 receptor activation in these microglial immune cells in the brain is working to break down those beta-amyloid plaques. And when you lose the P2Y2 receptor in that mouse model, those plaques develop quicker because the immune cells are no longer offering protection by chewing up that beta-amyloid. That’s one of the hypotheses we’re exploring right now.
Q: So you’d bet that these receptors are actually protective against Alzheimer’s?
JEAN: Yes. Going back to the human — it’s hard to get human tissues, especially brain tissues, but there is one published study that has shown that in Alzheimer’s patients who have passed away the P2Y2 receptor is down-regulated, meaning there’s not much left. Which would make sense. If it’s down-regulated, the plaques aren’t able to be chewed up, per se, by these microglia. There’s a correlation between low levels of P2Y2 receptors and Alzheimer’s disease that is apparent at the end of life.
LUKE: It’s very difficult to do some of these studies in humans because most of the available Alzheimer’s tissues are from end of life cases where you can only look at the end result of the disease without looking at the progression of the disease. Obviously you can’t take brain tissue from a living person, so the ability to study live cells from Alzheimer’s patients is limited. We rely very heavily on mouse models.
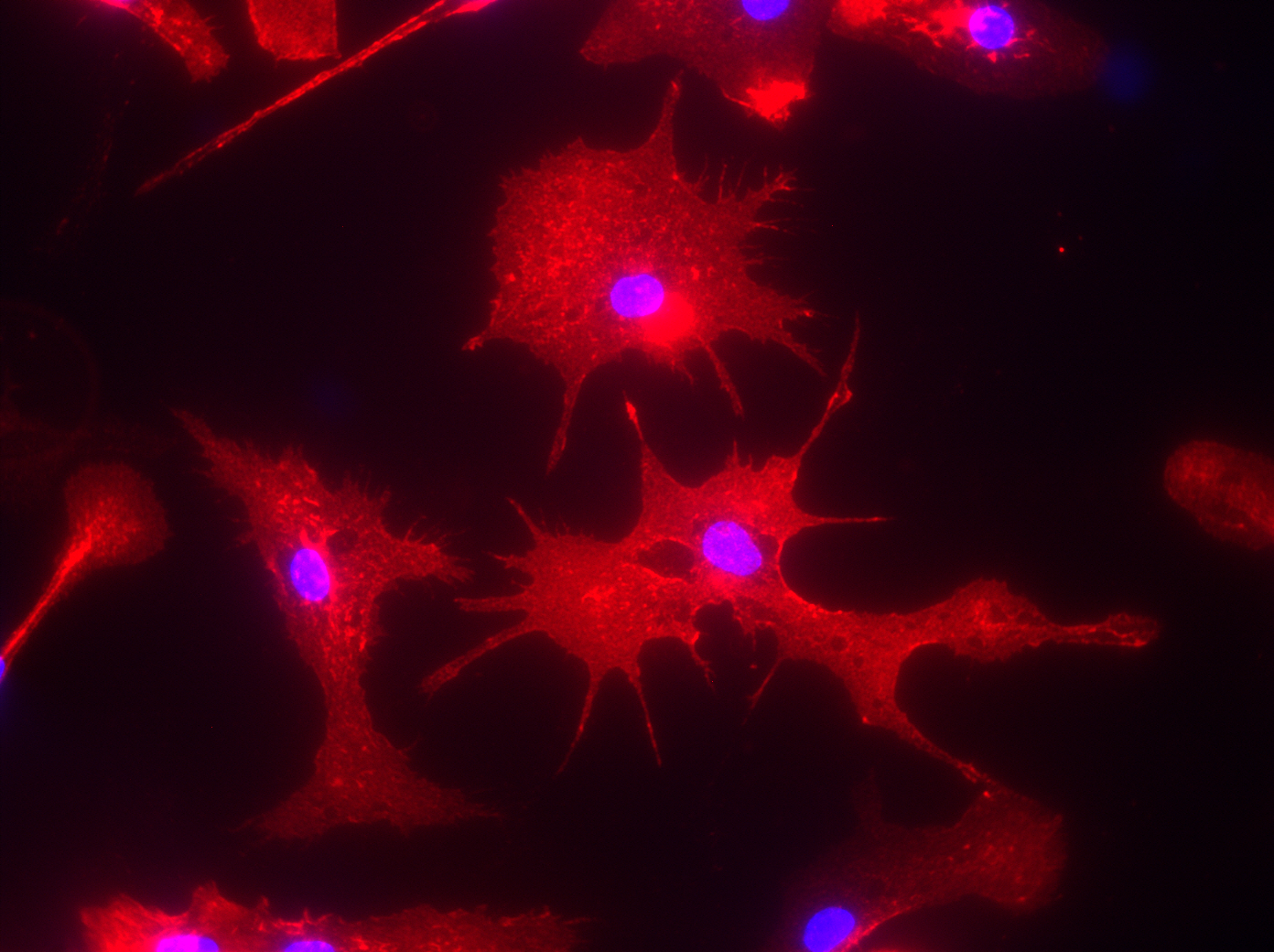
This immunofluorescence picture shows microglia cells that were isolated from the brain of an Alzheimer’s mouse model called TgCRND8 and cultured in a dish for further analysis. Image courtesy of Luke Woods.
Q: What have been the biggest shifts in our understanding of Alzheimer’s in recent years?
LUKE: Maybe one shift — I may not be the best expert to speak about it — is the idea that the beta-amyloid plaques are the cause of disease. It is now being mostly recognized that they’re really the tombstones of the disease. They’re not the initial cause, but rather the end result of the disease. For a long time investigators were focused on trying to prevent the buildup of beta-amyloid because that was one aspect of Alzheimer’s disease that you could see and measure. Now the thinking is that maybe the beta-amyloid does not contribute as much to disease progression as originally thought, and rather is the end result of a complicated mechanism that is actually causing the neurodegeneration.
JEAN: There’s still debate on what causes Alzheimer’s disease. There is a small percentage of patients where it’s actually related to a genetic alteration in the amyloid precursor protein gene.
LUKE: Another link has been with the ApoE (apolipoprotein E) gene, which makes a lipoprotein and cholesterol transporter. We inherit 1 copy of the ApoE gene from each of our parents and it has been shown that individuals who have at least 1 copy of a particular variant of the gene called ApoE4 are at increased risk of developing Alzheimer’s disease.
Q: From the perspective of a lab scientist, why do you care about Alzheimer’s?
JEAN: We care about any disease, really, and if we can show that our receptors have anything to do with any disease, we’d be proud to have a role in that.
LUKE: We don’t do much clinical science here, it’s mostly basic science. We contribute to the basic understanding of the disease so that drug companies and medicinal chemists who develop drugs for clinical use in Alzheimer’s patients can say, “Hey, this group’s research found a new mechanism related to Alzheimer’s disease, so let’s target this pathway to treat the disease.” It’s always nice to contribute to that sort of ground-level science.
JEAN: That would be the ideal, to show that whether you have to activate or inhibit the P2Y2 receptor, it does something to improve the clinical outcome in Alzheimer’s patients. A better understanding of Alzheimer’s and other diseases is what’s needed — we’re just working to provide a piece of the puzzle.
Q: How has being down in the trenches changed your perspective on research and Alzheimer’sin general?
JEAN: We’re the ones who are hoping to clarify the direction for science to go. We do the experiments and we are the first ones to see the data. We collect the data that becomes the cornerstone for deciding the direction our research goes. I think Gary would agree with that — he depends on us a lot to collect the data and we depend on him to help determine which scientific findings to chase and which ones not to chase.
I’ve been doing this for 35 years, and I really do enjoy the science. I’ve seen the science of these nucleotide receptors come a long way. These receptors have in common their use of extracellular nucleotides, particularly ATP (or adenosine triphosphate, more commonly known as the intracellular high energy molecule of all cells). And this ATP, is at a high concentration inside cells, so when it is released by cell damage, it can easily activate nucleotide receptors on nearby cells. It was Dr. Geoffrey Burnstock, now considered to be the grandfather of nucleotide receptors, who claimed a long time ago that there are receptors on the outside of cells that respond to ATP. Everybody kind of laughed at him, “Yeah, sure, right. There’s no way: ATP belongs inside the cell.” So for me personally, to come in on the ground level for these receptors and find a role for them in a variety of diseases has been exciting for me.
LUKE: ATP is the energy currency inside of all cells, so it’s use outside cells would be like tossing money out the window. Why would they want ATP outside the cell? It didn’t make any sense at the time, but looking back I think it does. What happens if you damage or rupture a bunch of cells during an injury? You get the release of a high concentration of ATP that neighboring cells recognize as a danger signal telling them that an injury has occurred. In that sense, ATP makes the perfect signaling molecule to tell other cells that an injury has occurred and they need to start the repair work by recruiting immune cells to the damaged tissue.
Jean Camden has spent 35 years working at the University of Missouri and more than a decade in Gary Weisman’s lab. // Photo by CALEB O’BRIEN/Bond LSC
Q: Where would you like to be in five years with this research?
JEAN: I talked about determining how the P2Y2 receptor in this mouse model was protective. It would be nice to find out which cell type on which the P2Y2 receptor is expressed in contributes most to neuroprotection. Our hypothesis would be that the microglial cells are very important, since they gobble up beta-amyloid, but other cell types including neurons and endothelial cells are likely involved. We’re also anxious to look at other inflammatory diseases to see if the P2Y2 receptor plays a similar role there.
LUKE: From somebody who does a lot of bench work, something I would like to see is a really good tool, a specific agonist or antagonist of the P2Y2 receptor that could be used in the clinic. There are a few suitable compounds available that we use to investigate the P2X7 receptor— I’ve told you that some have been tested in clinical trials — but the P2Y2 receptor has been sort of an enigma, due to the lack of selective inhibitors and agonists that are specific enough for clinical use. I’d like to see the development of a specific agonist or antagonist that could eventually be used to treat inflammatory diseases. There’s no reliable drug that is currently suitable to investigate the P2Y2 receptor in animals or humans, so clearly more work is needed there.
This interview has been edited for length and clarity.
Roger Meissen/Bond Life Sciences Center – These soybean roots show some nematode cysts. The small, white circles are the hardened body of the nematodes and form when the nematode attaches itself to the root to create a feeding cell.
Beneath a North Carolina field in 1954, a tiny worm inched its way through the soil and butted against a soybean root. The worm pierced the plant, slipped inside and inserted a needle-like appendage into a cell. It pumped a mixture of proteins into the root cell and waited for the potent blend to take effect on the unsuspecting soybean.
Since the first detection of soybean cyst nematode (SCN) in the US, the worm Heterodera glycines has spread to about 80 percent of American soybean fields. In Missouri, SCN attacks soybeans in almost every county and causes decreased yields even in robust, healthy-looking fields. Nationwide, SCN wreaks havoc to the tune of $1.2 billion per year, making it by far the most costly soybean pest.
Despite the hefty toll, farmers still depend on the same small handful of resistant soybean varieties to combat SCN that they have used for years. But those natural defenses are becoming less effective as nematodes evolve.
“More than 90 percent of the soybean cultivars that farmers plant derive their resistance from a single source,” said Melissa Mitchum, a plant nematologist at the University of Missouri Bond Life Sciences Center and Division of Plant Sciences faculty member in the College of Agriculture, Food and Natural Resources. “Consequently, this has led to widespread virulence in the pathogen population, thereby reducing the effectiveness of those resistant cultivars.”
But in the past 10 years, researchers studying SCN have made numerous breakthroughs, unlocking the secrets of the nematode and exploring how the worm interacts with host plants. Now, scientists are poised to bring that knowledge from the laboratory to the field.
Found in translation
Relatively little was known about SCN a decade ago.
Scientists could determine the type of nematode in a soil sample and had just figured out the cocktail of proteins a nematode pumps into the soy root cell that transform it into a syncytium, or feeding cell.
Working in part with funding from commodity boards and farmer checkoff dollars, researchers around the country made breakthrough after breakthrough, deepening our understanding of SCN and equipping scientists with new tools to fight the pest.
That money helped scientists sequence the soybean genome, draft a SCN genome and pinpoint important soy and SCN genes.
Checkoff investments continued to pay dividends in 2012 when Mitchum and colleagues cloned the first gene linked to natural soybean cyst nematode resistance. This breakthrough is one key step in moving science from the laboratory into the field. With a SCN resistance gene in hand, new avenues for creating soybean varieties that fight off the nematode are opening up.
But other areas of research also hold promise in the struggle against soybean cyst nematode’s parasitic ways.
Mitchum’s group also identified the plant receptors that recognize and respond to the blend of proteins an attacking nematode inserts into a plant. In a recent project published in Plant Biotechnology Journal, Xiaoli Guo, a postdoctoral fellow in Mitchum’s lab demonstrated that silencing those receptors in soybean roots helped the plant resist SCN.
This work has implications for more crops than just soybeans: Working with collaborator Xiaohong Wang at Cornell, Mitchum’s group used their understanding of plant receptors to develop a potato resistant to potato cyst nematode.
A roadmap for discovery
To build on the momentum of recent research, experts drafted a roadmap for the next decade of nematode research. Their goal, Mitchum said, is to address the challenge of translating these research breakthroughs into something tangible for the farmer.
With support from state farmer run organizations such as the Missouri Soybean Merchandising Council, the North Central Soybean Research Program and the United Soybean Board, researchers are formulating teams that “bring together commodity, industry and university funding to develop collaborative, interdisciplinary, multistate projects,” said Mitchum.
And there’s plenty of scientific firepower to advance research: MU’s College of Agriculture, Food and Natural Resources alone has more than 90 faculty studying plant science, plant genetics and other areas of agriculture-related science.
The scientists’ plan for the next 10 years involves a blend of molecular research, plant breeding, population biology and outreach. Researchers will focus on refining the existing draft SCN genome, which will help to develop a quick, inexpensive test for HG type and eventually contribute to understanding of how SCN overcomes a plant’s resistance. They’ll create an “atlas” of SCN genes researchers can use to block the pest. Updating yield loss estimates and mapping SCN distribution will also give scientists a better idea of the nematode’s national impact. Other efforts will allow breeders to incorporate new sources of resistance into commercially-available varieties, refine the use of non-host species to control SCN and develop a pipeline for creating and testing transgenic SCN-resistant soybeans. Finally, videos, webinars and training modules will help scientists, students and producers take advantage of new discoveries and techniques.
Roger Meissen/Bond Life Sciences Center – Michael Gardner, Ph.D. student, Melissa Mitchum, associate professor of Plant Sciences, Xiaoli Guo, post doctoral fellow, conduct research at the University of Missouri. They investigate how soybean cyst nematode overcomes soybean resistance to identify novel approaches for management.
Onward with research
A thorough understanding of SCN resistance and virulence starts with basic research and then moves into the field. “We all need to come together to transfer this knowledge to the breeder,” Mitchum said, “and from there it gets out to the farmer.”
Her lab recently received a National Science Foundation grant to continue their work on soybean protein receptors. Specific targeting of the receptors is just one potential strategy for producing new kinds of SCN-resistant plants. A second grant, from the National Institute of Food and Agriculture, will allow the lab to continue refining their understanding of how SCN proteins overcome a host plant’s defenses. To that end, Mitchum’s graduate student Michael Gardner is identifying the genetic blueprint of the different SCN types present in Missouri fields.
“If we better understand nematode populations and what makes those populations distinct, we can better advise farmers confronted with virulent nematodes,” Gardner said. “We’ll be able to go one step beyond the HG type test and understand how nematodes are able to adapt in the long term, not just the next growing season.”
But these breakthroughs do little good unless they then become useful tools for breeders and ultimately farmers. To that end, Mitchum and other researchers will help breeders use research results to produce soybeans with durable resistance. They‘ll also develop guides so farmers can easily incorporate new technologies and management strategies into their farms.
It’s important for farmers, breeders and researchers to take a unified approach to fighting SCN, Mitchum said, because a tactic that seems successful at first could backfire.
For instance, combining resistance genes in a single soybean variety could actually be harmful. “When we deploy it in the field, we select for nematodes that can overcome multiple types of resistance,” Mitchum said.
A better approach might be to perfect varieties with distinctive resistance mechanisms and insure durable resistance by rotating among the resistant varieties and non-host crops.
“It’s similar to taking antibiotics,” Mitchum said. “Improper use and overuse selects for resistance.” The strategic planning document should help everyone working with soybeans and SCN leverage and build upon new knowledge.
Despite all the research and recent breakthroughs, there remains only one certainty in the ongoing arms race between soybeans and SCN: “It is highly unlikely that we will eradicate it.” Mitchum said, “We’re going to have to find new strategies to protect and bolster soybean yields.”
Thanks to the efforts of researchers such as Mitchum, in the future SCN might be a little easier to get along with.
Roger Meissen/Bond Life Sciences Center – Research specialist and coordinator for the Plant Nematology Lab Amanda Howland processes soil samples for nematodes. Howland replaced Bob Heinz earlier this year.
University of Missouri Plant Nematology Laboratory: An extensive legacy
Bob Heinz spent his last day at work in December surrounded by nematodes. Heinz served as Mitchum’s research specialist and coordinator of the Plant Nematology Laboratory, where he processed soil samples, responded to growers and assisted researchers. After 35 years on the job, he’s retired, and Amanda Howland is now filling his shoes. The scientists and farmers who’ve worked with Bob over the decades thank him for his dedication and wish him luck in his retirement. And Amanda: Welcome aboard.
The Plant Nematology Lab, housed within Mitchum’s lab at MU, represents a successful model for how research, teaching and extension program integration can promote interdisciplinary collaboration. Such an approach helps maintain an effective pipeline that brings research-based information and resources from MU to Missourians. The lab offers an array of tests that help farmers understand and manage nematode populations. The available tests include:
–Vermiform Nematode Identification: Soybean Cyst? Root Knot? Lesion? Find out what kinds of nematodes are in your fields with this test.
–Soybean Cyst Nematode Egg Count: This procedure provides an estimate of the number of SCN eggs in your field.
–Soybean Cyst Nematode HG Type Test: Different types of SCN have overcome various sources of soybean resistance. A HG type test will help you determine the best source of resistance for the particular type of SCN in your field.
Epigenetics involves changes in how your genes work.
In classical genetics, traits pass from generation to generation in DNA, the strands of genetic material that encode your genes. Scientists thought alterations to the DNA itself was the only way changes could pass on to subsequent generations.
So say you lost a thumb to a angry snapping turtle: Because your DNA hasn’t changed, your children won’t be born with smaller thumbs. Classic.
Things get way more complicated with epigenetics. It turns out that some inherited changes pass on even though they are not caused by direct changes to your DNA. When cells divide, epigenetic changes can show up in the new cells.
Getting nibbled on by an irate turtle isn’t likely to epigenetic changes, but other factors such as exposure to chemicals and an unhealthy diet, could cause generation-spanning epigenetic changes.
How does it work?
The main players in epigenetics are histones and methyl groups.
Imagine your genes are like pages in a really long book. Prior to the mid-1800s, books came with uncut edges, so in order to read the book, you’d have to slice apart the uncut pages. That’s sort of what a histone does to DNA: They are proteins that wrap DNA around themselves like thread on a spool. They keep the DNA organized and help regulate genes.
Methyl groups (variations on CH3) attach to the histones and tell them what to do. These molecules are like notes in a book’s margin that say, “These next few pages are boring, so don’t bother cutting them open.” As you read the book, you’ll save time and effort by skipping some sections even though those sections still exist. Or maybe the note will say, “This next section is awesome; you’ll want to read it twice.”
That’s epigenetics. Higher level cues that tell you whether or not to read a gene.
And when a scribe makes a copy of the book, they’ll not only copy all the words in the novel, but all the other stuff, too: the stuck-together pages and the margin notes.
What about my health?
Many areas of health — including cancer, autoimmune disease, mental illness and diabetes — connect with epigenetic change.
For example, scientists link epigenetic changes to neurons to depression, drug addiction and schizophrenia. And environmental toxins — such as some metals and pesticides — can cause multigenerational epigenetic effects, according to research. Once scientists and doctors decipher those processes work, they will be better equipped to treat the sick and be able to take preventative measures to help insure our health and the health of our kids.
Is it epigenetics or epigenomics?
Confusing, I know.
As we learned from the first question, epigenetics is “the study of heritable changes in gene function that do not involve changes in the DNA sequence,” according to my trusty Merriam-Webster.
Epigenomics is the study and analysis of such changes to many genes in a whole cell or organism. It’s comparable to the difference between genetics (dealing with particular pieces of DNA, usually a gene) and genomics (involving the whole genetic shebang).
Where can I learn more?
Start at this year’s Life Sciences and Society Program Symposium, “The Epigenetics Revolution: Nature, Nurture and What Lies Ahead,” on March 13-15, 2015. Speakers from all over the country will delve into the puzzles and possibilities of epigenetics.
For more background, Nature magazine also created this supplement on epigenetics.
A yellow light indicates oxidant production in the tissue of a migrating fly larva. Source: Tobias Dick, German Cancer Research Center | Illustration by Paige Blankenbuehler
University of Missouri research characterizes a novel compound
By Paige Blankenbuehler | Bond LSC
Your body has an invisible enemy.
One that it creates all on it’s own called oxidative stress, long thought of as an underlying cause of some of humanity’s most insidious diseases – cancer, Alzheimer’s, Parkinson’s Disease, cardiovascular disease and diabetes.
Every day, our bodies are exposed to harmful free radicals known as reactive oxygen species as a result of our environment.
But, when something goes wrong with this energy extraction process, cells become inundated with reactive oxygen compounds that cause oxidative stress. The search for drugs to treat the problem have been ongoing, and with a complicated problem like oxidative stress, it’s all about finding the right combination.
Recent research by Bond Life Sciences Center investigator and Biochemistry Department Professor, Mark Hannink, provides a new approach for addressing the problem of oxidative stress and a starting point on developing a drug in pill form.
Mark Hannink and Kimberly Jasmer, a Ph.D. student in his lab, recently helped characterize a new molecule (called HPP-4382) that provides a novel way to treat oxidative stress. Their research was done in a partnership with High Point Pharmaceuticals, LLC, of North Carolina, where this new molecule was developed.
Oxidative stress can cause damage to the building blocks of a cell, resulting in excessive cell proliferation in the case of cancer or cell death in the case of neurodegenerative diseases like Parkinson’s.
Often, the majority of stressors are actually created inside our own cells as a byproduct of how our cells extract energy from the food that we eat and the air that we breathe.
Specimen of protein are prepared for an experiment in a lab at the Bond Life Sciences Center at the University of Missouri.
Understanding oxidative stress
Most simply, oxidative stress is an imbalance that happens when the body uses oxygen to produce energy.
Superoxide, a “promiscuous and nonspecific” compound produced as a byproduct of this process, is a highly reactive molecule that can damage DNA and other cellular components, Hannink said.
The superoxide molecule is a “free radical.” That means it’s especially promiscuous and reacts with many different types of cellular molecules, leaving destruction in its wake, he said.
That damage can lead to a long list of problems, including cancer or neurodegenerative diseases like Parkinson’s disease.
In response to oxidative stress, the cell produces protective “anti-oxidant” proteins, which help remove the harmful reactive oxygen species and minimize damage.
But a heavy anti-oxidant response could be dangerous, too. The bottom line: it’s about maintaining a fine balance between “oxidants” and “anti-oxidants”.
Search for right combination continues
A drug that corrects the imbalance of oxidative stress could one day have wide applicability.
Jasmer developed a test to measure how specific compounds altered gene expression. The genetic response to oxidative stress has both an “ON” switch and an “OFF” switch.
Using this test, Jasmer determined how each compound affected specific genetic switches and, in turn, how the response to oxidative stress is regulated.
This test helped identify which molecules might be promising candidates for treating oxidative stress, leading them to one in particular that seemed to have the desired properties: HPP-4382.
But creating effective drugs is a long process of trial and error. Once molecules have been identified that show efficacy in lab-based assays, scientists try different combinations to increase their potency and drug-like properties, and High Point is currently testing other molecules that behave like HPP-4382.
The compound serves as a good starting point for researchers who are interested in understanding how oxidative stress affects cellular processes, such as cell proliferation or cell death.“Now we have a better understanding of what this compound is doing,” Hannink said. “This compound can be used to test different ideas of how the balance between oxidants and anti-oxidants is achieved in healthy cells and how perturbation of this balance can lead to different diseases.”
Graduate students Yuleam Song and Dan Salamango inoculate a bacteria culture in Johnson’s lab. The inoculation takes a small portion of a virus and multiplies the sample, allowing researchers to custom-make viruses.
By Madison Knapp | Bond Life Sciences Center summer intern
Modern science has found a way to turn viruses —tiny, dangerous weapons responsible for runny noses, crippling stomach pains and worldwide epidemics such as AIDS— into a tool.
Gene therapy centers on the idea that scientists can hijack viruses and use them as vehicles to deliver DNA to organs in the body that are missing important genes, but the understanding of virus behavior is far from exhaustive.
Marc Johnson, researcher at the Christopher S. Bond Life Sciences Center and associate professor of molecular microbiology and immunology in the MU School of Medicine, has been building an understanding of viral navigation mechanisms which allow a virus to recognize the kind of cell it can infect.
Johnson’s research specifically explores the intricacies of the viral navigation system and could improve future direction of gene therapy, he said.
Marc Johnson (left) with Dan Salamango, a graduate student that works in his lab. The lab does important research on the basic function and mechanisms of viral navigation and transport.
Turning a virus into a tool
Conceptualized in the 1970s, gene therapy was developed to treat patients for a variety of diseases, including Parkinson’s, leukemia and hemophilia (a genetic condition that stops blood from clotting).
To treat disease using gene therapy, a customized virus is prepared. A virus can be thought of as a missile with a navigation system and two other basic subunits: A capsule that holds the ammunition and the ammunition itself.
The viral genetic material can be thought of as the missile’s ammunition. When a cell is infected, this genetic material is deployed and incorporated into the cell’s DNA. The host cell then becomes a factory producing parts of the virus. Those parts assemble inside the cell to make a new virus, which then leaves the cell to infect another.
The capsule is made of structural protein that contains the genetic material, and the navigation system is a protein that allows the virus to recognize the kind of cell it can infect.
Viral navigation
Gene therapy uses viruses to solve many problems by utilizing a virus’ ability to integrate itself into a host cell’s DNA; to do this successfully, researchers need to provide a compatible navigation component.
In the body, viruses speed around as if on a busy highway. Each virus has a navigation system telling it which cells to infect. But sometimes if a virus picks up the wrong type of navigation system, it doesn’t know where to go at all.
“What you can do is find a virus that infects the liver already, steal its navigation protein and use that to assemble the virus you want to deliver the gene the liver needs,” Johnson said. “You can basically take the guidance system off of one and stick it onto another to custom design your virus.”
But this doesn’t always work because of incompatibility among certain viruses, he said.
Johnson and his lab are working to understand what makes switching out navigation proteins possible and why some viruses’ navigation systems are incompatible with other viruses.
“I’m trying to understand what makes it compatible so that hopefully down the road we can intelligently make others compatible,” Johnson said.
The right map, the right destination
Johnson creates custom viruses by introducing the three viral components—structural protein, genetic material, and navigation protein—to a cell culture. The structural protein and genetic material match, but the navigation component is the wild card. It could either take to the other parts to produce an infectious virus, or it could be incompatible.
Johnson uses a special fluorescent microscope to identify which viruses assembled correctly and which didn’t.
A successful pairing is like making a match. If a navigation protein is programmed to target liver cells, it’s considered a successful pairing when the virus arrives at the liver cell target location.
The scope of gene therapy continues to widen. Improved mechanisms for gene therapy, and greater knowledge of how a navigation protein drives a virus could help more people benefit from the vehicles viruses can become.
Johnson uses several high-profile model retroviruses, including human immunodeficiency virus (HIV), which affects an estimated 35 million people worldwide each year, according to the World Health Organization.
Understanding nuances of HIV in comparison to other viruses allows Johnson to pick out which behaviors might be common to all retroviruses and others behaviors that might be specific to each virus.
Johnson said his more general approach makes it easier to understand more complex viral features.
“If there are multiple mechanisms at work, it gets a little trickier,” Johnson said. “My angle is more generic, which makes it easier to tease them apart.”