Female rats struggle to find their way in BPA study from MU and the NCTR/FDA
Cheryl Rosenfeld is one of 12 researchers partnering with the NCTR/FDA to study BPA
Despite concerns about bisphenol A (BPA), academic and regulatory scientists have yet to reach a consensus on BPA’s safety.
The National Institute of Environmental Health Sciences (NIEHS), the National Toxicology Program (NTP), the Food and Drug Administration and independent university researchers are working together to change that.
“The idea of this Consortium is to examine the potential systems that have been previously suggested to be affected by BPA,” said Cheryl Rosenfeld, an associate professor of biomedical sciences at the University of Missouri and one of twelve researchers involved in the project.
Rosenfeld’s group looked at spatial navigation learning and memory. They found that prenatal exposure to BPA could potentially hinder the ability of female rats to learn to find their way through a maze. This effect was not seen in male rats.
Approved by the FDA in the early 1960s, BPA can be found in a wide variety of products, including plastic food and drink containers with recycle codes 3 or 7, water and baby bottles, toys, the linings of metal cans and water pipes, even patient blood and urine samples.
BPA has structural similarities to estrogen and can potentially act as a weak estrogen in the body.
In Rosenfeld’s experiment, researchers at the National Center for Toxicology Research gave pregnant rats a fixed dose of BPA every day: a low, medium, or high dose.
After the baby rats were born, researchers continued to dose the babies, both male and female, according to what their mothers had received.
When these rats reached three months old, they were tested in a circular maze with twenty possible exit holes, one of which was designated as the correct escape hole. Every day for seven days, researchers tested the rats’ abilities to solve the maze in five minutes and timed them as they ran.
Rats solve mazes in three ways, Rosenfeld said.
They can run through the labyrinth in a spiral pattern, hugging the outer walls, and work their way in until they find the correct exit hole in what is called a serial search strategy.
Or they might move aimlessly in the maze using an indirect search strategy, Rosenfeld said. “In this case, the rats seemingly find the correct escape hole by random chance.”
Lastly, they can travel directly from the center of the maze to the correct escape hole. The third strategy is considered the most efficient method because the rats find their way swiftly, Rosenfeld said.
Sarah Johnson, a graduate student and first author on the paper, assessed each rat’s performance in the maze using a three-point tracking program that recognizes the rat’s nose, body, and tail.
Using the program, Johnson measured their performances in terms of the total distance traveled, the speed at which the rat ran the maze, how long it took the rats to solve the maze (latency), and how often the rat sniffed at an incorrect hole.
The last two parameters are considered the best gauges of spatial navigation learning and memory.
“What you expect to see is that they should start learning where that correct escape hole is,” Rosenfeld said. “Thus, their latency and sniffing incorrect holes should decrease over time.”
Rosenfeld’s group found that female rats that had been exposed to the highest dose of BPA since fetal development were less likely to find the escape hole than rats that hadn’t been exposed to BPA.
As for how this study may translate to people, Rosenfeld said, “the same brain regions control identical behaviors in rodents and humans.”
She considers it a starting point for setting up future experiments that take into consideration sex differences in cognitive behaviors and neurological responses to BPA.
Immediate next steps for the Rosenfeld group include analyzing tissue collected from the brains of rats that had undergone maze testing. Rosenfeld’s team of researchers will measure DNA methylation and RNA expression in the brain to determine which genes might be involved in navigational learning and memory. Their overarching goal is to determine how changes in observed sex- and dose-dependent behaviors occur on the molecular level.
NIEHS grant U01 ES020929 supported this research. Additional coauthors include Mark Ellersieck and Angela Javurek of the University of Missouri, Thomas H. Welsh Jr. of Texas A&M University, and Sherry Ferguson, Sherry Lewis, and Michelle Vanlandingham of the National Center of Toxicological Research/Food and Drug Administration. Read the full study on the Hormones and Behavior website and browse the supplementary data for this work.
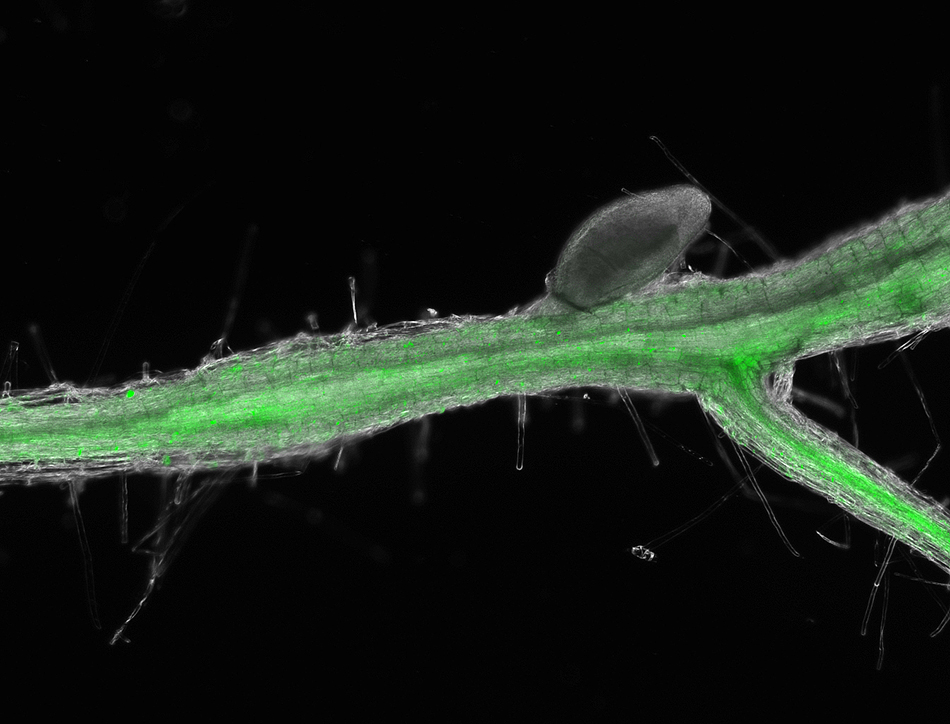
Scientists find how nematodes use key hormones to take over root cells
This Arabidopsis root shows how the beet cyst nematode activates cytokinin signaling in syncytium 10 days after infection. The root fluoresces green when the TCSn gene associated with cytokinin activation is turned on because it is fused with a jellyfish protein that acts as a reporter signal. Contributed by Carola De La Torre
Roger Meissen | Bond Life Sciences Center
This is a story about spit.
Not just any spit, but the saliva of cyst nematodes, a parasite that literally sucks away billions in profits from soybean and other crops every year.
Researchers are working to uncover exactly how these tiny worms trick plant root cells into feeding them for life.
Cytokinin is normally produced in plants, but these researchers determined that this growth hormone is also produced by nematode parasites that use it to take over plant root cells.
“While it’s well-known that certain bacteria and some fungi can produce and secrete cytokinin to cause disease, it’s not normal for an animal to do this,” said Melissa Mitchum, an MU plant scientist and co-author on the study. “This is the first study to demonstrate the ability of an animal to synthesize and secrete cytokinin for parasitism.”
Not Science Fiction
Reprogramming another organism might sound like a far out concept, but it’s a reality for plants susceptible to nematodes.
Cyst nematodes hatch from eggs laid in fields and quickly migrate to the roots of nearby plants. They inject nematode spit into a single host cell of soybean, beet and other crop roots.
Carola De La Torre
“Imagine a hollow needle at the head of the nematode that the parasite uses to penetrate into the plant cell wall and secrete pathogenic proteins and hormone mimics,” said Carola De La Torre, a co-author of the study and plant sciences PhD student with Mitchum’s lab. “Nematodes use the spit to transform the host cell into a nutrient sink from which they feed on during their entire life cycle. This de novo differentiation process greatly depends on nematode–derived plant hormone mimics or manipulation of plant hormonal pathways caused by effector proteins present in the nematode spit.”
These effector proteins and other small molecules in their spit cause the root cell to forego normal processes and create a huge feeding site called a syncytium. In a short period of time, this causes hundreds of root cells to combine into a large nutrient storage unit that the nematode feeds from for its entire life.
Being able to convince a root cell to do the nematode’s bidding starts with a takeover of the plant host cell cycle — which regulates DNA replication and division. This implies that a plant hormone like cytokinin is involved, says Mitchum. Cytokinin normally regulates a plant’s shoot growth, leaf aging, and other cell processes.
Proving the relationship
While Mitchum’s lab had a hunch that cytokinin was key to this takeover, proving it took some creative science.
De La Torre and Demosthenis Chronis, a postdoctoral fellow MU at the Bond LSC depended on mutant Arabidopsis plants to explore the relationship. “One of the great things about using Arabidopsis as our host plant is the vast genetic resources of cytokinin and hormone mutants that are available through the scientific community,” De La Torre said.
She infected Arabidopsis that contained a reporter gene called TCSn/GFP with nematodes. This gene is associated with cytokinin responses within the plant cells and is fused with a jellyfish protein that glows green when turned on. So, De La Torre saw nematodes activated cytokinin responses in the plant early after infection when her plants emitted a green fluorescent glow under the microscope.
Next, she infected plants missing the majority of their cytokinin receptors with nematodes. Then she started counting nematodes present.
“After a careful evaluation of nematode infection, we observed less female nematodes developing in the receptor mutants compared to the wild type” De La Torre said. “The nematodes could not infect well, and that was a clear piece of evidence suggesting that cytokinin plays a main role in plant–nematode interactions.”
Another experiment looked at Arabidopsis containing a reporter gene called GUS that was fused to the regulatory sequences of the cytokinin receptor genes. All three cytokinin receptor genes were activated where the nematode was feeding.
A final experiment used a mutant that created an excess of an enzyme that degrades cytokinin, finding that a base level of plant cytokinin was also necessary for nematode growth.
“The simple statement is that the cytokinin receptors were activated in response to nematode infection and the mutants did not support growth and development of the nematodes,” Mitchum said. “This shows that if you take away the ability of the plant to recognize cytokinin the worms are unable to fully develop.”
An international collaboration
Mitchum’s team did not work alone.
The lab of Florian Grundler at Rheinische Friedrich-Wilhelms-University of Bonn, Germany, was also on a mission to uncover if genes in the nematode controlled cytokinin activation. They had identified a key gene in the beet cyst nematode that makes the cytokinin hormone. When they took away the ability of the nematode to secrete cytokinin certain cell cycle genes were not activated at the feeding site and the nematodes did not develop. Now we know that the nematode is also secreting cytokinin to modulate the pathways.
De La Torre took that information and found the same gene in the soybean cyst nematode.
Now, Mitchum’s team is trying to find how this key gene might work differently in other nematode types, like root-knot nematode as part of a new National Science Foundation grant. They hope this will help lead to better resistance in future crops.
“Understanding how the nematode modulates its host is going to help us exploit new technologies to engineer plants with enhanced resistance to this terribly devastating pathogen,” Mitchum said. “Technology is changing all the time, we’re gaining new tools constantly, so you never know when something new is going to allow us to do something specific at the site of nematode feeding that will lead to a breakthrough.”
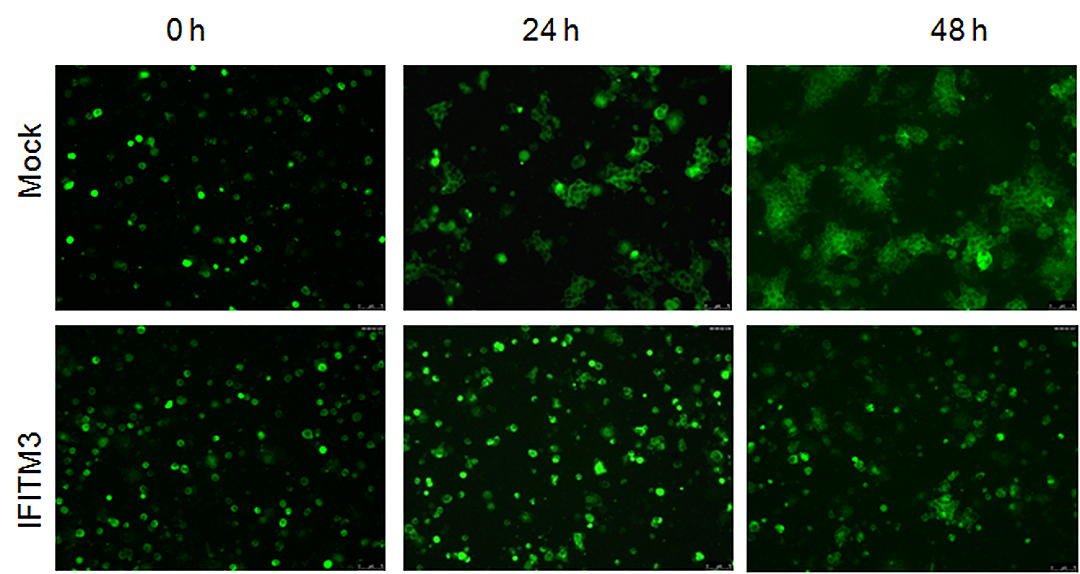
Cells that expressed IFITM proteins (bottom row), showed much less spread of HIV-1 compared with cells lacking the protein. | courtesy Jordan Wilkins, Liu Lab
By Jennifer Lu | MU Bond Life Sciences Center
For Shan-Lu Liu, thinking outside the box meant putting an antiviral protein inside HIV-infected cells, rather than into healthy ones.
Liu and his team of researchers studied how interferon-induced transmembrane (IFITM) proteins limit the infection of HIV-1, the primary strain of virus responsible for AIDS. The journal Cell Reports published their results on September 17.
IFITM proteins are biomolecules with broad antiviral properties. Although multiple versions of IFITM have been found in humans, three are known to have antiviral properties: IFITM1, IFITM2 and IFITM3.
In a 2013 paper published in PLoS Pathogens, the Liu laboratory demonstrated that these three IFITM proteins have the ability to thwart a variety of viral infections.
Shan-Lu Liu, Bond Life Sciences scientist and associate professor in the MU School of Medicine department of molecular microbiology and immunology. | Photo by Justin Kelley, University of Missouri Health Care
“They can inhibit influenza virus, Ebolavirus, HIV and SARS coronavirus,” said Liu, an associate professor in the Department of Molecular Microbiology and Immunology at the Bond Life Sciences Center.
Liu wanted to know why IFITM’s inhibition of HIV was uncharacteristically weaker than its inhibition of other viruses.
To study this conundrum, many researchers designed their experiments by expressing IFITM proteins in target or healthy cells. Then they infected these IFITM-bolstered cells with HIV, but saw minimal protection against viral infection.
In a twist, the Liu group put IFITM proteins in HIV-1 producer or infected cells instead of in healthy T-lymphocyte cells, a special kind of immune cell used specifically to study the viral infection by HIV.
They found that IFITM proteins, especially IFITM2 and IFITM3, interacted with the viral envelope protein (Env) that makes up the outer shells of virus particles.
For normal HIV infections to occur, Liu said, envelope proteins need to be cleaved into two parts.
Once processed, the resulting two portions, Env gp120 and gp41, can be incorporated into viral particles. The two processed envelope proteins protrude from the outer surface of the virus like mushroom-shaped pegs that help the virus latch on and fuse to target cells.
But when IFITM binds to envelope proteins they interfere with the viral envelope functions.
“It’s just unexpected,” Liu said, about this finding. In other viruses his group has studied, IFITM inhibited virus’ ability to fuse its outer shell with the membrane of a cell by making the cell membrane rigid during the infection process. He said he assumed IFITM would block HIV the same way.
Instead, they found evidence suggesting that IFITM blocks infection through direct contact with HIV’s envelope proteins.
“It is the first study that shows this kind of interaction,” Liu said. “That’s why this study is so surprising. We did not think about this.”
Liu does not yet know the mechanism behind IFITM and envelope protein interactions, but he said the outcome remains clear. “IFITM proteins inhibit this Env cleavage process and this makes HIV less infectious and less transmissible.”
To visualize IFITM’s inhibitory effects in action, Liu’s group tagged HIV-1 inside infected cells with a green fluorescent dye. Then they colored healthy target cells with a red fluorescent dye. When they mixed the two populations of cells together, they saw two days later a very tiny amount of cells exhibiting green signals within the red cells —a sign of spread of HIV cell-to-cell infection.
By comparison, cells in the control group—healthy red-tagged cells mixed with green HIV-infected cells that do not contain IFITM—showed a higher number of red cells lighting up green inside.
This suggested that having IFITM and HIV-1 inside the virus-producing cells somehow limited the virus’ infectivity and cell-to-cell spread at the same time.
His group also showed through a technique called co-immunoprecipitation that IFITM proteins bound specifically with envelope protein rather than with other proteins, such as Gag. Liu attributed this work to his two talented hardworking graduate students, Jingyou Yu and Minghua Li.
Unfortunately, the benefits of IFITM are short-lived. When the Liu laboratory let HIV-infected cells replicate again and again, they saw that HIV could evolve enough to circumvent the inhibitory effects of IFITM after 30 passages.
Liu said that his research on IFITM was still in its early stages, but that the next step would be to look at the IFITM’s function in HIV patients in order to move the basic research of IFITM from bench to bedside.
“Once we know better how this protein works, we can develop some inhibitors to block HIV, block Ebola, block other viruses,” Liu said. “So that’s our ultimate goal.”
Liu is a Bond Life Sciences Center investigator and an associate professor of molecular microbiology and immunology in the MU School of Medicine. He studies how viruses infect healthy host cells to cause illness and cell response to viral attacks.
The National Institutes of Health and the Canadian Institutes of Health Research partially supported this research. Additional collaborators include Eric Freed, PhD, senior investigator with the National Cancer Institute (NCI) HIV Dynamics and Replication Program; Chen Liang, PhD, at McGill University; and Benjamin K. Chen, PHD, at the Mount Sinai School of Medicine. Read the full study on the Cell Reports website and browse the supplementary data for this work. See more on this research from Mizzou News.
Endocrine disruptors impact physical activity and metabolism in mice
By Caleb O’Brien | MU Bond Life Sciences Center
Could your experiences in the womb make you lazy as an adult?
A recent study of California mice suggests that early exposure to environmental chemicals can later impact an animal’s metabolism and level of voluntary physical activity, according to new University of Missouri research.
“We found that if we developmentally exposed California mice to bisphenol A (BPA) or ethinyl estradiol (EE), the estrogen present in birth control pills, it caused later disruptions in voluntary physical activity,” said Cheryl Rosenfeld, a researcher in MU’s Bond Life Sciences Center and associate professor of biomedical sciences in the College of Veterinary Medicine. “What that means is they move around less in their home cage, they’re more likely to sleep, and they engage in less voluntary physical activity.”
Rosenfeld’s lab studies the ways that exposure to environmental chemicals such as BPA can affect other behaviors, including cognition and parenting. Endocrine-disrupting chemicals can accumulate in the environment and act like the hormones naturally produced by many organisms, including humans. To test the chemicals’ impact on metabolism and activity, the lab used California mice. This mouse model is a good model for metabolic diseases. And because these animals are initially derived from the wild, they may better replicate the genetic diversity of most human populations.
The researchers exposed the mice to BPA and EE in the womb and until weaning via the mom’s diet. A third group of mice whose mothers were placed on a phytoestrogen-free control diet was not exposed to either chemical. The scientists then placed all the mice on this same control diet and measured their energy expenditure, body composition and level of voluntary physical activity as adults.
To test those attributes, Rosenfeld’s lab relied on a variety of tools and techniques. They rigged bicycle computers to “hamster wheels” to track how far, fast and for how long the mice ran. Using a device called a “Promethion continuous measurement indirect calorimetry system” the researchers continuously monitored the mice’s energy expenditure by measuring oxygen consumption and carbon dioxide production and by using a three-beam system, tracked the rodents’ movements during the dark and light cycles.
Later, the researcher measured the animals’ body composition using an EchoMRI, a tiny MRI machine the size of a filing cabinet, and finally measured circulating concentrations of glucose and hormones that regulate metabolism.
Female mice exposed to BPA and EE were less active than control mice. They moved around their cages less at night (when the nocturnal California mouse is considered most active), moved more slowly, drank less water, and spent more time sleeping. In addition, BPA-exposed females burned more carbohydrates relative to fats, as compared to control mice. This is similar to the difference between obese and slender humans, and many researchers believe that burning more carbohydrates relative to fats can lead to fats gradually accumulating in the body.
“It’s worrisome that environmental chemicals we are exposed to in utero can override our genes and disrupt our neuro-circuitry,” said Sarah Johnson, a research specialist and graduate student in Rosenfeld’s lab and primary author on the study. “The net effect is that we can have behavioral disruptions into adulthood, including altered physical activity.”
The researchers are currently conducting follow-up studies to determine if the changes caused by exposure to BPA and EE predispose mice to obesity and other metabolic disorders. They also are interested in exploring if exposure could affect the children and grandchildren of these mice and examining the potential underlying neural mechanisms.
“Our findings are significant because decreased voluntary physical activity, or lack of exercise, can predispose animals or humans to cardiovascular diseases, metabolic disorders and even cancer,” Rosenfeld said.
Other authors on the study are Angela Javurek and Michele Painter (MU Biomedical Sciences), Mark Ellersieck (MU Agriculture Experimental Station- Statistics), Charles Wiedmeyer (MU Veterinary Medical Diagnostic Laboratory and Department of Veterinary Pathobiology) and John Thyfault (Kansas University Medical Center, Molecular and Integrative Physiology)
The study, “Sex-Dependent Effects of Developmental Exposure to Bisphenol A and Ethinyl Estradiol on Metabolic Parameters and Voluntary Physical Activity” was supported by NIH Grant 5R21ES023150 (to C.S.R.) and R01DK088940 (JPT) and was published in the Journal of Developmental Origins of Health and Disease.
James Amos-Landgraf, assistant professor of comparative medicine and genetics at the University of Missouri, is helping develop a pig model for colon cancer using CRISPR. //photo by CALEB O’BRIEN/Bond LSC
James Amos-Landgraf needed a pig.
The assistant professor of comparative medicine and genetics at the University of Missouri had joined forces with a startup company developing a tool to detect early colon cancer-causing lesions. They already tried out a rat-sized model, but still needed a full-sized prototype.
Scientists in Europe had an ideal pig model for colon cancer, but importing the animals presented a problem. It would be prohibitively expensive and time consuming, and the method European scientists used to develop the pig took several years and cost a great many Euros, Amos-Landgraf said.
Those obstacles might have been enough to scuttle the project entirely, but CRISPR, a new gene-editing tool discovered in the DNA of a peculiar bacterium, has changed the equation for scientists everywhere.
So when Amos-Landgraf went to the National Swine Resource and Research Center (NSRRC) to ask about importing pigs, they told him, “‘We can just make you the model,’” Amos-Landgraf said. “‘We should be able to do a CRISPR project within a few months.’”
CRISPR is rapidly reshaping the way biologist around the world do their jobs.
At Mizzou, it’s transforming how researchers learn about viruses and mosquitoes, pigs and zebrafish, and the individual genes affecting development, sickness and health. The tool makes research more efficient, cost-effective and vastly more powerful.
Amos-Landgraf knows firsthand just how time-consuming and laborious generating an animal model was pre-CRISPR.
“What was almost a two-year process just to generate an animal now would take us a matter of months,” Amos-Landgraf said. “I think the CRISPR revolution is going to be amazing for all of science. I’m totally intrigued by everything that’s going on with this.”
Borrowing a bacterial relic
CRISPR rolls off the tongue far more readily than its unabbreviated equivalent: “clustered regularly interspaced short palindromic repeats.” The name refers to a strange pattern scientists at the University of California, Berkeley noticed in the genome of a bacterium that lives in acidic, abandoned mines: groups of palindromic bacterial DNA sequences interspersed with segments of viral DNA.
It turned out that the genetic snippets were relics of the bacteria’s prior run-ins with viral invaders, like genetic mug shots on a most-wanted list.
Viruses are tiny packages of genetic material that hijack cells, such as bacteria, in order to reproduce. And when a virus enters a bacterial cell, the host compares the virus’ genetic material to the snapshots preserved in the bacteria’s own DNA. If they match, the bacteria dispatches a bounty-hunter protein called Cas9, which tracks down the virus and slices its DNA in half at the very spot that matched the virus’ genetic fingerprint.
If an unfamiliar virus attacks and the bacterium survives, it will incorporate a segment of the invader’s DNA into its own, adding a new battle scar to its DNA and a new miscreant to the most-wanted list.
When the researchers studying the bacterial immune system figured out how it worked, they realized the process could have implications far beyond the organism’s acidic abode: It could become a powerful, inexpensive, and versatile gene-editing tool.
A ground shift
The journey to better manipulate genes has been a long one.
For decades, scientists relied on various techniques and tricks to tease out the function of genes. The most common tool is forward genetics, where a researcher starts with an interesting characteristic in an organism and then hunts for the gene that caused it. Those characteristics could be traits that occur naturally, such as genetic diseases in purebred dogs or pigmentation in corn kernels, or a scientist could induce defects — essentially altering an organism’s genome by exposing it to a bath of nasty chemicals.
Anand Chandrasekhar, Bond Life Sciences Center biologist and professor in the division of biological sciences, and PhD candidate Suman Gurung take a look at some zebrafish they’ve modified using CRISPR. //photo by CALEB O’BRIEN/Bond LSC
Imagine that an organism is like a car, suggests Anand Chandrasekhar, a Bond Life Sciences Center biologist and professor in the division of biological sciences.
“You take a car that is running nicely and you have some kind of weird mechanic from Hell come in and mess something up — just one thing — and the car doesn’t run. Then you have to figure out why the car doesn’t run by looking carefully for where the defect is.”
Reverse genetics — unsurprisingly — starts on the other end. Researchers pick a gene of interest and try to silence or alter it. If they succeed, then they look for changes in the organism that suggest the altered gene plays a role in the observed characteristic.
This tool shaped how scientists do research and what animals they use in their labs. In fact, model organisms such as mice rose in popularity partly because of how easily reverse genetic techniques like homologous recombination work with them, said Amos-Landgraf. But this approach was time consuming, expensive and didn’t function well on other organisms.
The next step forward were Zinc-Finger Nucleases (ZFNs) and Transcription Activator-Like Effector Nucleases (TALENs). Both act like guided missiles to strike at a gene of interest, targeting a specific region of genetic material and breaking both strands of the organism’s DNA at that spot. Once the DNA is broken, the cell’s natural repair mechanism intervenes and stitches the gene back together.
However, the process is prone to errors — mutations — that can alter or silence a gene. ZFNs and TALENs reliably worked in a broader array of species.
CRISPRs represent the next advancement in this process and is far faster than previous techniques.
“Let’s say if you had 15 or 20 genes that you wanted to study: You could design a CRISPR reagent for each one of them in a couple of afternoons, whereas in the ‘olden days’ (three or four years ago) with TALENs that could have taken you months,” Chandrasekhar said. “And if you were using ZFNS… you would not even imagine doing it, because you would have been crazy.”
Elizabeth Bryda, a professor of veterinary pathobiology at MU who heads the Rat Resource and Research Center, stands by a Genetic Analyzer machine. //photo by CALEB O’BRIEN/Bond LSC
At MU’s NIH-funded Rat Resource and Research Center (RRRC), scientists think CRISPR will help break dependence on default model organisms. The RRRC is the only center of its kind in the US and one of two in the world, serving as a repository and distribution center for rats that model human diseases.
“We’re always preaching, use the species that’s most appropriate for the question you’re asking,” said Elizabeth Bryda, a professor of veterinary pathobiology at MU who heads the RRRC. “If you’re studying human disease, use the species that best recapitulates that disease. I think CRISPR will give people the flexibility to really work in the species they want to be working in.”
For example, the center is using CRISPR to develop rat models of human inflammatory bowel diseases, such as Crohn’s disease. “All of those barriers to making rat models are no longer issues,” Bryda said, “CRISPR is easy and finally allows us to manipulate rats in ways we haven’t before.”
That’s good news for the RRRC: “I do think we’re going to see a huge increase in the number of rat models,” Bryda said, “which would increase our inventory.”
Seeing through the zebrafish
Zebrafish are another model organism that might become even more important thanks to CRISPR.
Originally found in the rice paddies and streams of India and Myanmar, the minnow-like fish is an important model organism. They’re easy to care for, produce abundant offspring and — because their embryos are transparent — make great tools for studying development.
Chandrasekhar uses zebrafish to study cranial motor neurons, the neurons that connect to, and control, muscles in the head. His lab is especially interested in the way those cranial motor neurons are deployed during development: how the neurons know where to go and to which muscles they should link.
“CRISPR is a really big boon for research, because now even small labs can test tens of genes over a short period of time for their effect on a particular biological process,” Chandrasekhar said. “That’s how we use it: We study the process of cell migration within a nervous system, and we want to study a whole slew of associated genes.”
Researchers have identified hundreds of new and potentially important genes using advance genomics, but the old techniques of reverse genetics were too slow and tedious to keep up with the new discoveries.
“CRISPR has removed the bottleneck,” Chandrasekhar said. “We can rapidly go through and, hopefully, find new genes and new signaling pathways that might be playing a very specific role for the migration and the biological process that we study.”
But finding a new gene is just the beginning, Chandrasekhar said.
“We have one student who is testing five genes, and if even one or two of those genes turn out to be important, that will then be sufficient for the lab to continue working on them for two or three years.”
Although scientists primarily use CRISPR as Chandrasekhar does, to silence genes in model organisms, new genes can also be introduced.
Through a process called homologous directed repair, scientists select a location where they want to introduce a gene and design a CRISPR to target that region.
Daniel Davis, a PhD candidate and lab manager for Assistant Professor of Veterinary Pathobiology Catherine Hagan, is developing a technique to screen potential antidepressant drugs by leveraging CRISPR technology and the advantages of the zebrafish. //photo by CALEB O’BRIEN/Bond LSC
Daniel Davis, a PhD candidate and lab manager for Assistant Professor of Veterinary Pathobiology Catherine Hagan, is developing a technique to screen potential antidepressant drugs by leveraging CRISPR technology and the advantages of the zebrafish.
When a zebrafish is stressed, it produces a neurotoxic compound, but when the fish is calm, it produces a different compound, one that is neuro-protective. The difference depends on which key enzyme the fish produces — in a stressful situation, the fish produces more of the enzyme that leads to neurotoxicity.
Davis is using CRISPR to try to link different fluorescent proteins genes to each branch of this stress pathway: If the fish produces more of the stressful compound, it will also produce a red fluorescent protein. If the other pathway is taken, the fish will assemble green fluorescent protein.
“If you take some fish, subject them to a stressor and test a variety of potential therapeutics on them, you could visualize the fluorescent proteins to see which therapeutics are more protective,” Davis said.
Altering the host to understand the virus
Other models present special challenges. In mosquitoes, for instance, it’s hard to knock out genes from its genome using traditional methods.
“The problem is that in mosquitoes such as Aedes aegypti, ‘traditional’ knockouts never really worked, so people tried out new techniques such as ZFNs and TALENs,” said Alexander Franz, assistant professor of veterinary pathobiology at MU. But the other techniques had flaws, too: they were expensive, complicated to assemble and often posed issues of efficiency and specificity.
Franz studies arthropod-borne viruses (arboviruses), specifically dengue virus and chikungunya virus. The life cycle of an arbovirus requires its circulation between arthropods, such as mosquitoes, and vertebrate hosts, such as humans. Because vaccines exist for only a few mosquito-borne viruses — yellow fever and Japanese encephalitis, for example — people usually rely on conventional and often ineffective environmental controls to thwart disease: bed nets, the elimination of breeding areas, insecticides.
Alexander Franz (far right), an assistant professor of veterinary pathobiology at MU who demonstrated that CRISPR is effective in mosquitoes, stands with his lab. From left to right: Velmurugan Balaraman, Asher Kantor, Hannah Gerlt, and Shengzhang Dong. //Photo courtesy of Alexander Franz
Franz is pursuing a different avenue for protection that uses genetic manipulations to interrupt the transmission cycle of a virus in the mosquito.
“If you can stop the virus from taking hold in the mosquito, you can block transmission of the virus to its vertebrate host,” he said. “But to do so, you need an effective way to manipulate the mosquito’s genome.”
This is where CRISPR comes into play. “When people started reporting using the CRISPR system for genome editing in Drosophila or zebrafish, we immediately had the idea to try it out in mosquitoes.” Working with two postdocs, Franz demonstrated for the first time that the CRISPR system was capable of disrupting genes in mosquitoes.
To do so, he started with a line of transgenic mosquitoes that had already been modified to produce red and blue fluorescent proteins in their eyes. The lab designed a CRISPR to silence the gene responsible for the blue fluorescent protein. After trying a few different methods, they found a technique that turned off the target gene when they injected the CRISPR into mosquito embryos.
Because it is a very powerful and easy-to-handle genome editing technique, CRISPR has been recently utilized and further developed by other groups studying mosquito-pathogen interactions.
Other MU researchers focus on the viral interaction with human host cells.
Marc Johnson, associate professor of molecular microbiology and immunology at the Bond Life Sciences Center, studies the way a virus puts itself together inside a host cell and fights off the cell’s defenses. //photo by CALEB O’BRIEN/Bond LSC
Marc Johnson, associate professor of molecular microbiology and immunology at the Bond Life Sciences Center, studies the way a virus puts itself together inside a host cell and fights off the cell’s defenses.
“We don’t know all the cellular genes, cellular machinery and cellular pathways that viruses are harnessing,” Johnson said. “The best way to say that a virus requires a particular gene would be to knock it out of the cell and see if the virus can still replicate.”
“CRISPR is a real ground shift in how we can do science,” he said. “Things that took 6 months to a year to make one gene before, now we can do half-a-dozen in a week.”
The technique has altered the rate at which Johnson’s research proceeds and expanded the scope of his lab’s work. “It’s allowed me to take a step back and think about the whole genome, as opposed to being totally focused on this one thing or that one thing,” Johnson said. “I’d never really taken a step back to think about the whole genome — every gene, where are they and what families. It’s changed my outlook on the cell, the way I can think about it.”
The CRISPR era
Amos-Landgraf and the researchers at NSRRC are still in the process of validating their pig model: developing primers to identify the mutation and creating the CRISPRs themselves. Once everything is ready, they’ll test out the lesion-detecting colonoscope, and if all goes well, move into human trials — far faster and more economically than would have been possible a few years ago.
But Amos-Landgraf is tantalized by the possibilities the technology offers beyond increased speed and reduced costs: “To be able to tease apart not just a single gene in a pathway, but maybe think about knocking out or altering all the genes in a pathway and looking at combinations of those pathways… You can start thinking about multiple gene knockouts, multiple gene manipulations all within the same experiment,” he said.
“And that is not only cost saving, but it becomes a really powerful tool when you want to interrogate biology. We’ve entered a new era of genetics and genomics.”
Tommy Langdon waits for a bee to land on a flower. // photo by CALEB O’BRIEN/BondLSC
Emily Fulcher came face-to-face with science while dissecting a hackberry gall: “Ewww,” she exclaimed, “it’s peeking out a little bit!”
Fulcher and 12 other high school students were observing plant galls as part of a Summers @ Mizzou camp exploring “The Arts as a Portal to Science Communication.”
The camp, which ran Sunday through Thursday afternoon, was facilitated by three interdisciplinary scholars and artists adept at bringing together science and the arts: Milbre Burch, an award-winning storyteller, poet and artist; Lee Ann Woolery, leader of the MU Extension Community Arts Program; and Suzanne Burgoyne, director of MU’s Center for Applied Theatre and Drama Research.
The goal of the camp was “to strengthen interdisciplinary research,” Woolery said, and to encourage “imaginative, creative minds from different disciplines to work together.”
Heidi Appel and Jack Schultz explain the complicated, contentious relationship between plants and insects during the Summers @ Mizzou camp “The Arts as a Portal to Science Communication.” // photo by CALEB O’BRIEN/BondLSC
On Monday, Heidi Appel, co-director of the Schultz-Appel Chemical Ecology Lab, and Jack Schultz, director of the Bond Life Sciences Center, were invited to give the students a crash-course on the complex realm of plant-insect interactions and the equally nuanced world of scientific research. They delved into the nature and process of science as a knowledge-generating enterprise: How do we know what we know? What is peer review? Why do grants matter?
After the talk, the students split into groups — half dissected galls and scrutinized their contents under the microscope, while the other group ventured out into the Tucker greenhouse and to the tiny prairie patch next door, where they recorded the sounds of insects pollinating and eating plants. And they visited the lab of Rex Cocroft, professor of biological sciences at MU, where they used lasers to record the vibrations produced when caterpillars munch on Arabidopsis leaves.
Drawings and collages, such as this illustration of a gall by Emily Fulcher, lined the entrance to Ellis Auditorium, where students gave presentations on the the final day of the Summers @ Mizzou camp “The Arts as a Portal to Science Communication.” //photo by CALEB O’BRIEN/BondLSC
The rest of the week, the students explored different artistic media and disciplines to communicate science and deepen their own understanding of research. They painted, wrote, created videos and performed.
On the final afternoon of the camp, they gathered in Ellis Auditorium with the other Summers @ Mizzou camp participants, facilitators and family members for a final presentation. The arts and sciences group read brief, poetic explanations of the research they’d encountered earlier in the week, accompanied by amusing theatrical interpretations.
For Appel, the benefits of the camp go both ways: “Explaining my work to non-scientists makes me a better scientist because I have to clarify and distill my work down to a few key points,” Appel said. Illuminating her work for others affords an opportunity to step back and take a fresh look at the broader context of her research.
Cameron Christensen, a fifteen-year-old student from Jefferson City, said the camp offered a unique opportunity to learn how to convey information in different formats. The challenge of their final presentation, for example, was to “take cold data and make it lifelike,” Christensen said.
Appel, who has worked with all three of the camp’s facilitators in the past, said the camp “helps students understand that science is more than just a collection of facts.”
Cameron Christensen listens to the dulcet vibrations produced by a bug on a plant during the Summers @ Mizzou camp “The Arts as a portal to Science Communication.” // photo by CALEB O’BRIEN/BondLSC
Communicating science is increasingly recognized as a key step in the process of conducting and disseminating research, and invoking the arts can be a valuable tool in that process.
But in a way, putting the arts back in the sciences is just a long-overdue return to a rich heritage of creative polymathy, Woolery said. In the days before ubiquitous photography, keen powers of observation and adroit draftsmanship were de rigueur for anyone doing research — witness Audubon’s exquisite watercolors of America’s birds or Santiago Ramón y Cajal’s Miró-esque drawings of neurons.
For her part, Appel sees deep parallels between the arts and sciences.
“The processes themselves are not that different,” Appel said. “We all make observations and construct mental models. Then we either test a model (if we’re scientists), or we figure out a way to depict that model if we’re artists. Hypothesis testing is peculiar to science, but the other components are not that different.”
On the final day of the Summers @ Mizzou camp “The Arts as a Portal to Science Communication,” the students performed theatrical and literary interpretations of the science they studied. //photo by CALEB O’BRIEN/BondLSC
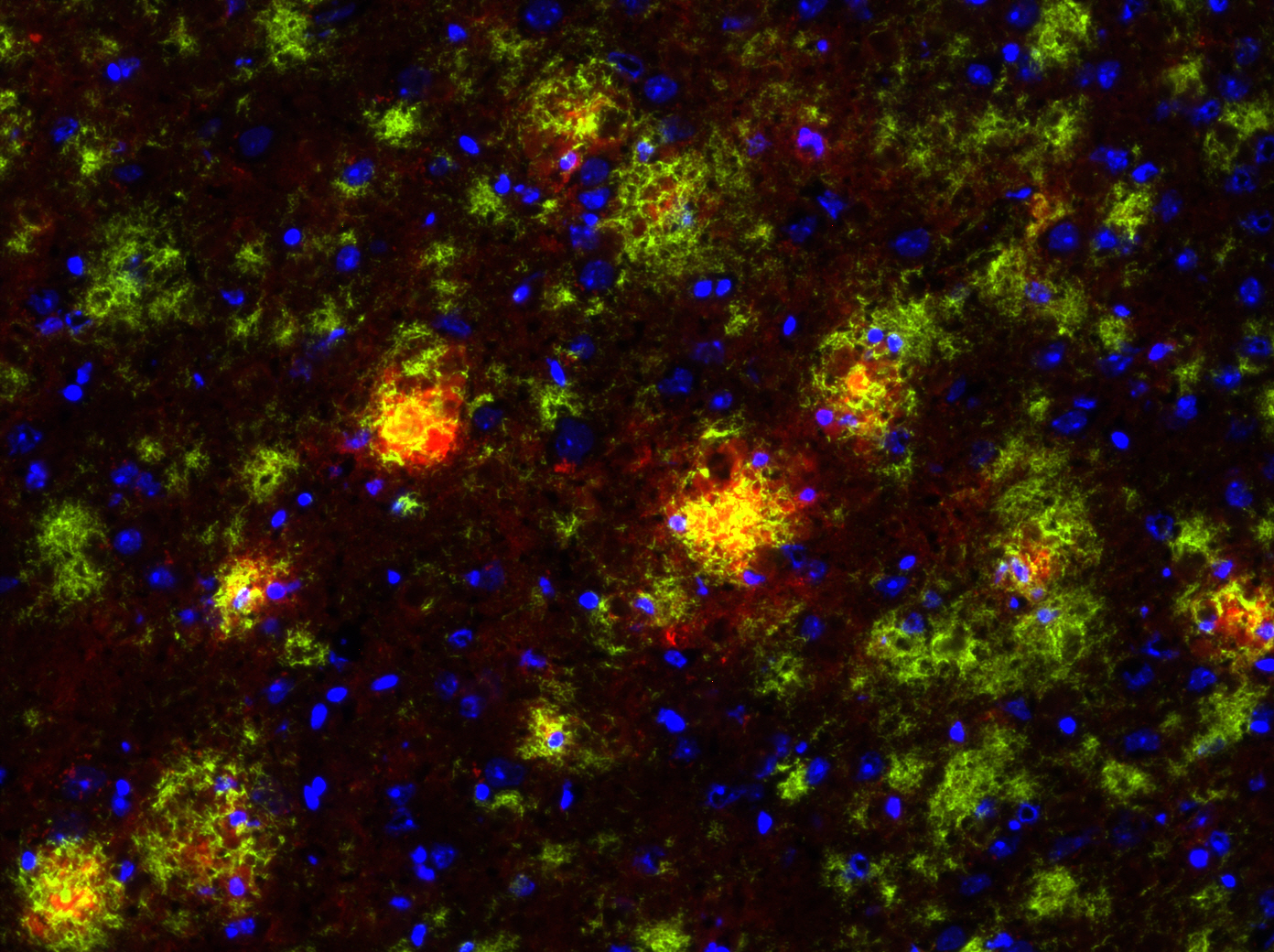
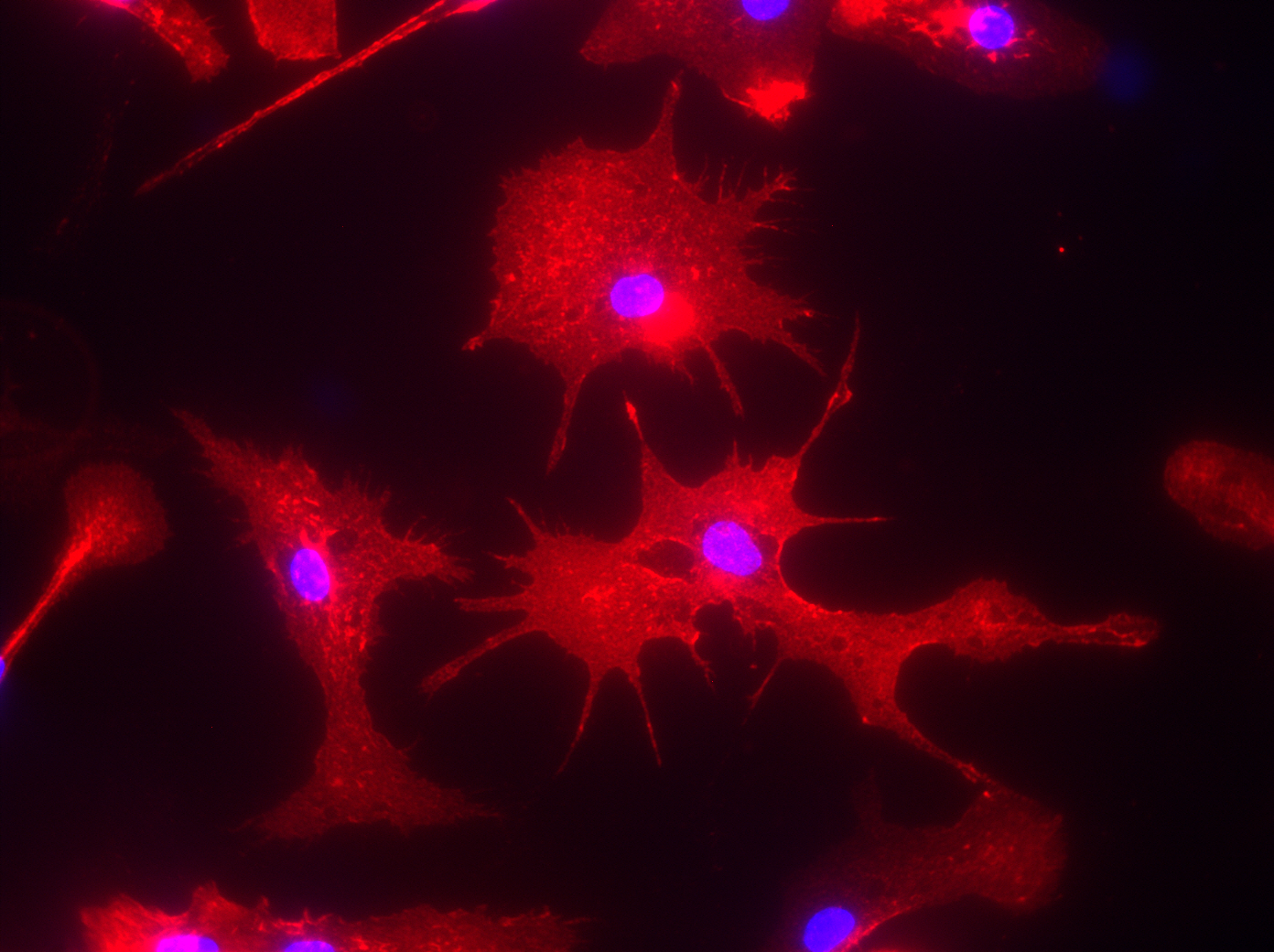
This immunofluorescence picture shows the brain of an Alzheimer’s disease mouse model, also known as the TgCRND8 mouse. In the picture, the amyloid beta plaques are stained green and the microglia, or immune cells of the brain, are stained red. Image courtesy of Luke Woods.
By Caleb O’Brien | MU Bond Life Sciences Center
Jean Camden and Luke Woods have an ant’s-eye view of Alzheimer’s disease.
Both are bench scientists in the laboratory of Gary Weisman, a professor of biochemistry at the Bond Life Sciences Center. Jean has spent the past 12 of her 35 years at the University of Missouri in the Weisman lab, running experiments, managing the lab and working with students. Luke joined the Weisman lab six years ago, doing what he call’s the dirty work of science: “Gary does the writing and the NIH stuff, but down in the trenches — that’s me and Jean.”
Weisman’s lab studies Alzheimer’s and other diseases, so I sat down recently with Jean and Luke to talk about their research for Alzheimer’s & Brain Awareness Month.
Q: What does your lab do, and how does it involve Alzheimer’s?
Luke: We primarily have two projects. One, which has been a very longstanding project, is focused on salivary glands and salivary gland inflammation. The other is the Alzheimer’s project. The link between them is a particular type of cell surface receptor called a nucleotide receptor — more specifically, a P2 nucleotide receptor called P2Y2. These P2 receptors function in a lot of different ways, but the link is with inflammation: We look at P2 receptors in salivary gland inflammation and in Alzheimer’s disease, which has a very large inflammation component that often gets glossed over. In a lot of Alzheimer’s articles that the public reads, you hear about amyloid beta plaques and tau tangles and neurodegeneration, but a large component of that is inflammation, where some of the resident non-neurons in the brain start responding like there’s inflammation in the brain, and it actually kills neurons. That’s been the focus in Gary’s lab for the past 30-plus years.
JEAN: The P2 receptors — especially the P2X7 and P2Y2 which we focus on — Gary during his postdoctoral work started studying these receptors without really knowing that they existed. At the time, he just knew that there was a pore formed in cells caused by the addition of the nucleotide ATP which eventually leads to apoptosis (cell death). Eventually, we cloned the human P2Y2 receptor gene with another group in North Carolina, so we call it “our receptor.” It only appears in cells under inflammatory conditions, such as Alzheimer’s disease, salivary gland autoimmune disease and cardiovascular disease. Any time you have tissue damage, it looks like the P2Y2 receptor is up-regulated. And then once the damage is healed, the receptor goes away.
Inflammation is good — we want inflammation, that’s how we heal — it’s the chronic inflammation that’s bad. But we really don’t know how these receptors work and what their role is during chronic inflammation. Do we want to activate them, or do we want to inhibit them?
LUKE: Scientists have investigated P2X7 receptor antagonists in the treatment of Crohn’s disease and rheumatoid arthritis — there are several clinical trials that have been focused on these receptors, evaluating whether you want to block or activate them. If you block them, you prevent the acute inflammatory responses that are good for wound healing; if you activate them, you may extend those responses past the healing phase into a chronic inflammatory phase that can be quite damaging. So unraveling that fine line of what you want to be doing to these receptors in disease settings is sort of what we do here.
Jean Camden and Luke Woods look at images of a mouse brain with Alzheimer’s disease. // Photo by CALEB O’BRIEN/Bond LSC
Q: When I think of Alzheimer’s, I think of a shriveled, shrunken brain, but I associate inflammation with swelling. Why the difference?
LUKE: I think the distinction is acute versus chronic inflammation. With acute inflammation, you get swelling. The body has different types of immune responses: acute responders like neutrophils and macrophages are immune cells that act quickly. They come in, for example, if you have a scratch, and there can be swelling. Along with macrophages neutrophils can protect cells from bacteria. The macrophages can also clean up damaged tissue and then the repair cells go to work. Cells come in that lay down a new matrix, whereas undamaged cells then migrate onto the matrix and regenerate. Well, what happens after you’re done repairing is that there are signals that tell the inflammation to stop. In chronic inflammation, that’s where you have continued cell death, and the tissue would then shrivel up. The shriveled brain that you’re referring to is during chronic inflammation, and that’s an end-of-life case, after a very long bout with Alzheimer’s.
JEAN: What we think of as inflammation is often a cut or a wound. It’s only been in recent years that Alzheimer’s disease has been considered an inflammatory disease. We have a phenomenal immune system, but when it goes awry, you have problems. In the other disease we look at — an autoimmune disease — your immune system starts to attack your own body. It’s hard to treat and understand the underlying mechanism.
Q: So how are you trying to unravel the role of inflammation in Alzheimer’s?
JEAN: To study Alzheimer’s, we have an Alzheimer’s mouse model. It overexpresses a gene for the amyloid precursor protein that enables the brain to accumulate high amounts of beta-amyloid plaques that you always hear about. So we’re using this mouse model that we’ve crossed with a mouse that does not express any P2Y2 receptor, so it’s called a knockout mouse. The P2Y2 receptor knockout mouse by itself is fine, and the Alzheimer’s mouse does develop Ab plaques, but it lives to approximately 6 months old before it will develop behavioral defects. The interesting thing is that when we cross the P2Y2 receptor knockout mouse with the Alzheimer’s mouse, the offspring that are Alzheimer’s mice without P2Y2 receptors prematurely die. So at least in this Alzheimer’s mouse model, it looks like the presence of the P2Y2 receptor is protective, because without it, the Alzheimer’s mice die much earlier. But we don’t really know which cell type is most important: Is it the P2Y2 receptor up-regulated on neurons that acts to repair them —which we’ve already shown happens — or is it the P2Y2 receptor on microglia (an immune cell of the brain), or is it the P2Y2 receptor on blood vessels in the brain that help recruit immune cells from the cardiovascular system to help with repair?
So we’re using this mouse model to investigate the role of the P2Y2 receptor, plus we also use cell lines because we can easily control the environment for these cell lines in culture. We isolate primary neurons, we can prepare primary microglial cells or we can purchase cell lines that comprise blood vessels. We can then utilize these tools to investigate cell signaling mechanisms for the P2Y2 receptor in individual cell types.
LUKE: One of the findings that we have found interesting in these primary cells is when you take them fresh out of the mouse, put them in a dish and then treat them as you wish. We’ve shown that if you activate the P2Y2 receptor in primary microglia from the mouse, they will actually engulf and chew up beta-amyloid. And so one of the things we think might be happening in this Alzheimer’s mouse model is that P2Y2 receptor activation in these microglial immune cells in the brain is working to break down those beta-amyloid plaques. And when you lose the P2Y2 receptor in that mouse model, those plaques develop quicker because the immune cells are no longer offering protection by chewing up that beta-amyloid. That’s one of the hypotheses we’re exploring right now.
Q: So you’d bet that these receptors are actually protective against Alzheimer’s?
JEAN: Yes. Going back to the human — it’s hard to get human tissues, especially brain tissues, but there is one published study that has shown that in Alzheimer’s patients who have passed away the P2Y2 receptor is down-regulated, meaning there’s not much left. Which would make sense. If it’s down-regulated, the plaques aren’t able to be chewed up, per se, by these microglia. There’s a correlation between low levels of P2Y2 receptors and Alzheimer’s disease that is apparent at the end of life.
LUKE: It’s very difficult to do some of these studies in humans because most of the available Alzheimer’s tissues are from end of life cases where you can only look at the end result of the disease without looking at the progression of the disease. Obviously you can’t take brain tissue from a living person, so the ability to study live cells from Alzheimer’s patients is limited. We rely very heavily on mouse models.
This immunofluorescence picture shows microglia cells that were isolated from the brain of an Alzheimer’s mouse model called TgCRND8 and cultured in a dish for further analysis. Image courtesy of Luke Woods.
Q: What have been the biggest shifts in our understanding of Alzheimer’s in recent years?
LUKE: Maybe one shift — I may not be the best expert to speak about it — is the idea that the beta-amyloid plaques are the cause of disease. It is now being mostly recognized that they’re really the tombstones of the disease. They’re not the initial cause, but rather the end result of the disease. For a long time investigators were focused on trying to prevent the buildup of beta-amyloid because that was one aspect of Alzheimer’s disease that you could see and measure. Now the thinking is that maybe the beta-amyloid does not contribute as much to disease progression as originally thought, and rather is the end result of a complicated mechanism that is actually causing the neurodegeneration.
JEAN: There’s still debate on what causes Alzheimer’s disease. There is a small percentage of patients where it’s actually related to a genetic alteration in the amyloid precursor protein gene.
LUKE: Another link has been with the ApoE (apolipoprotein E) gene, which makes a lipoprotein and cholesterol transporter. We inherit 1 copy of the ApoE gene from each of our parents and it has been shown that individuals who have at least 1 copy of a particular variant of the gene called ApoE4 are at increased risk of developing Alzheimer’s disease.
Q: From the perspective of a lab scientist, why do you care about Alzheimer’s?
JEAN: We care about any disease, really, and if we can show that our receptors have anything to do with any disease, we’d be proud to have a role in that.
LUKE: We don’t do much clinical science here, it’s mostly basic science. We contribute to the basic understanding of the disease so that drug companies and medicinal chemists who develop drugs for clinical use in Alzheimer’s patients can say, “Hey, this group’s research found a new mechanism related to Alzheimer’s disease, so let’s target this pathway to treat the disease.” It’s always nice to contribute to that sort of ground-level science.
JEAN: That would be the ideal, to show that whether you have to activate or inhibit the P2Y2 receptor, it does something to improve the clinical outcome in Alzheimer’s patients. A better understanding of Alzheimer’s and other diseases is what’s needed — we’re just working to provide a piece of the puzzle.
Q: How has being down in the trenches changed your perspective on research and Alzheimer’sin general?
JEAN: We’re the ones who are hoping to clarify the direction for science to go. We do the experiments and we are the first ones to see the data. We collect the data that becomes the cornerstone for deciding the direction our research goes. I think Gary would agree with that — he depends on us a lot to collect the data and we depend on him to help determine which scientific findings to chase and which ones not to chase.
I’ve been doing this for 35 years, and I really do enjoy the science. I’ve seen the science of these nucleotide receptors come a long way. These receptors have in common their use of extracellular nucleotides, particularly ATP (or adenosine triphosphate, more commonly known as the intracellular high energy molecule of all cells). And this ATP, is at a high concentration inside cells, so when it is released by cell damage, it can easily activate nucleotide receptors on nearby cells. It was Dr. Geoffrey Burnstock, now considered to be the grandfather of nucleotide receptors, who claimed a long time ago that there are receptors on the outside of cells that respond to ATP. Everybody kind of laughed at him, “Yeah, sure, right. There’s no way: ATP belongs inside the cell.” So for me personally, to come in on the ground level for these receptors and find a role for them in a variety of diseases has been exciting for me.
LUKE: ATP is the energy currency inside of all cells, so it’s use outside cells would be like tossing money out the window. Why would they want ATP outside the cell? It didn’t make any sense at the time, but looking back I think it does. What happens if you damage or rupture a bunch of cells during an injury? You get the release of a high concentration of ATP that neighboring cells recognize as a danger signal telling them that an injury has occurred. In that sense, ATP makes the perfect signaling molecule to tell other cells that an injury has occurred and they need to start the repair work by recruiting immune cells to the damaged tissue.
Jean Camden has spent 35 years working at the University of Missouri and more than a decade in Gary Weisman’s lab. // Photo by CALEB O’BRIEN/Bond LSC
Q: Where would you like to be in five years with this research?
JEAN: I talked about determining how the P2Y2 receptor in this mouse model was protective. It would be nice to find out which cell type on which the P2Y2 receptor is expressed in contributes most to neuroprotection. Our hypothesis would be that the microglial cells are very important, since they gobble up beta-amyloid, but other cell types including neurons and endothelial cells are likely involved. We’re also anxious to look at other inflammatory diseases to see if the P2Y2 receptor plays a similar role there.
LUKE: From somebody who does a lot of bench work, something I would like to see is a really good tool, a specific agonist or antagonist of the P2Y2 receptor that could be used in the clinic. There are a few suitable compounds available that we use to investigate the P2X7 receptor— I’ve told you that some have been tested in clinical trials — but the P2Y2 receptor has been sort of an enigma, due to the lack of selective inhibitors and agonists that are specific enough for clinical use. I’d like to see the development of a specific agonist or antagonist that could eventually be used to treat inflammatory diseases. There’s no reliable drug that is currently suitable to investigate the P2Y2 receptor in animals or humans, so clearly more work is needed there.
This interview has been edited for length and clarity.
The next time you slather mustard on your hotdog or horseradish on your bun, thank caterpillars and brassica for that extra flavor.
While these condiments might be tasty to you, the mustard oils that create their flavors are the result of millions of years of plants playing defense against pests. But at the same time, clever insects like cabbage butterflies worked to counter these defenses, which then started an arms race between the plants and insects.
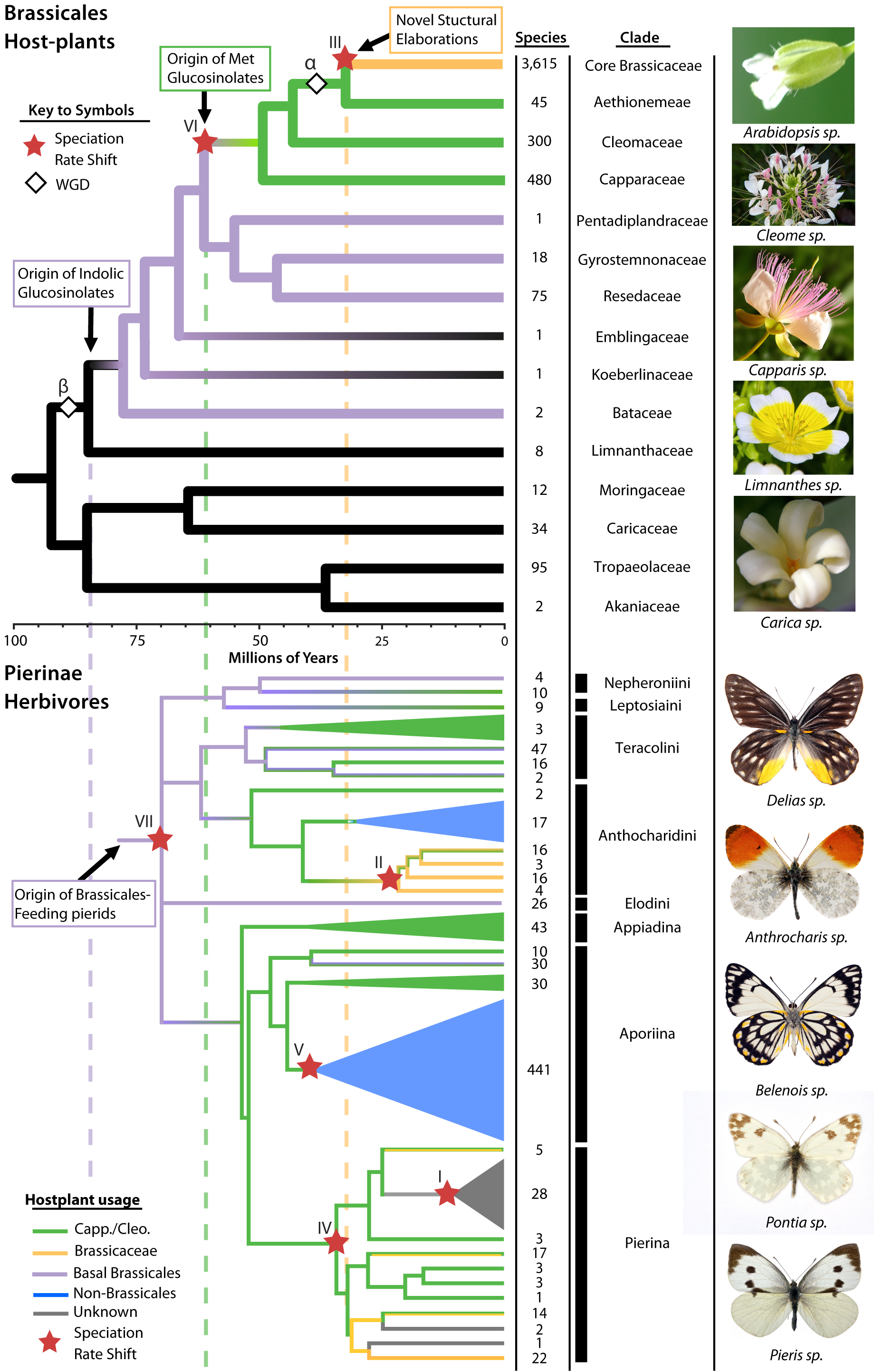
An international research team led by University of Missouri Bond Life Sciences Center researchers recently gained insight into a genetic basis for this co-evolution between butterflies and plants in Brassicales, an order of plants in the mustard family that includes cabbage, broccoli and kale.
“We found the genetic evidence for an arms race between plants like mustards, cabbage and broccoli and insects like cabbage butterflies,” said Chris Pires, an MU Bond Life Sciences Center researcher and associate professor of biological sciences in the College of Arts and Sciences. “These plants duplicated their genome and those multiple copies of genes evolved new traits like these chemical defenses and then cabbage butterflies responded by evolving new ways to fight against them.”
A biting taste
While you might like the zing in mustard, insects don’t.
Compounds, called glucosinolates, create these sharp flavors in plants to defend against caterpillars, butterflies and other pests. Brassicales species first evolved glucosinolate defenses around the KT Boundary — when dinosaurs went extinct — and eventually diversified to synthesize more than 120 different types of this compound.
For most insects, these glucosinolates prove toxic, but certain ones like the cabbage butterfly evolved ways to detoxify the compounds.
“Seeing the variation in the detoxification mechanisms among species and their gene copies gave us important evolutionary insights,” said Hanna Heidel-Fischer, a lead author on the study based at the Max Plank Institute for Chemical Ecology in Germany.
To look at these genetic differences, the team used 9 existing Brassicales genomes and also generated transcriptomes — the set of all RNA in a cell — across 14 Brassicales families. This allowed the team to map an evolutionary family tree of sorts over the millennia, seeing where major defense changes occurred. This family tree was compared with the family tree of 9 key species of Pieridae butterflies, which includes the cabbage butterfly.
Pires and his colleagues identified three significant evolutionary waves over 80 million years, where plants developed defenses and insects evolved counter tactics.
Pat Edger | Image by Roger Meissen, Bond LSC
“We found that the origin of brand-new chemicals in the plant arose through gene duplications that encode novel functions rather than single mutations,” said Pat Edger, a former MU post doc and lead author on the study. “Given sufficient amounts of time the insects repeatedly developed counter defenses and adaptations to these new plant defenses.”
This back-and-forth pressure resulted in the evolution of many more species of plants and butterflies than in other groups without glucosinolate pressures.
Proving an old concept
Co-evolution is not a new idea.
About 50 years ago two now-renowned biologists, Peter Raven and Paul Erhlich, introduced the idea of co-evolution to science. Using cabbage butterflies and Brassica plants as a prime example, the two published a landmark study in 1964 advancing the idea that two species can mutually influence the development and evolution of each other.
“Using Ehrlich and Raven’s principles and models, we looked at the evolutionary histories of these plants and butterflies side-by-side and discovered that major advances in the chemical defenses of the plants were followed by butterflies evolving counter-tactics that allowed them to keep eating these plants,” Wheat said.
Chris Pires and colleagues mapped the evolution of Brassicales and butterflies to find how each evolved to combat the defenses of the other. | Courtesy Chris Pires
This research provides striking support for the ideas of Ehrlich and Raven published 50 years ago.
“We looked at the patterns 50 years ago, and found conclusions that proved correct,” said Peter Raven, professor emeritus of the Missouri Botanical Garden and a former University of Missouri Curator. “The wonderful array of molecular and other analytical tools applied now under leadership of people like Chris Pires, provide verification and new insights that couldn’t even have been imagined then.”
Understanding more about how plants and insects co-evolve could one day lead to advances in crops.
“If we can harness the power of genetics and determine what causes these copies of genes, we could produce plants that are more pest-resistant to insects that are co-evolving with them—it could open different avenues for creating plants and food that are more efficiently grown,” said Pires.
Proceedings of the National Academy of Sciences (PNAS) published the study, “The butterfly plant arms-race escalated by gene and genome duplications,” in June. The National Science Foundation (PGRP 1202793), the Knut and Alice Wallenberg Foundation and the Academy of Finland provided the funding for this research.
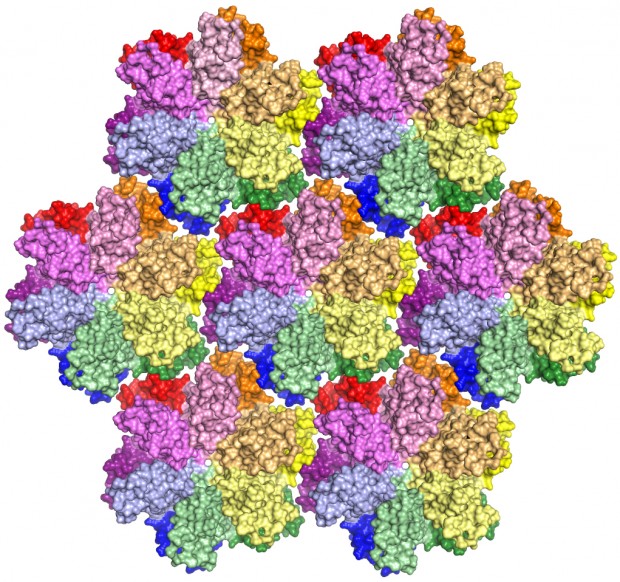
The HIV capsid protein (shown above in an array of hexagons) plays a critical role in the virus life cycle. Bond LSC researchers recently developed the most complete model yet of this vital protein. Image by Karen Kirby and Anna Gres
Seeing the whole picture can mean a lot when it comes to figuring out HIV.
Researchers at the University of Missouri Bond Life Sciences Center are gaining a clearer idea of what a key protein in HIV looks like, which will help explain the flexible protein’s vital role in the virus life cycle.
“The capsid acts as an invisibility cloak that hides the virus’ genetic information, the genome, while it is being copied in a hostile environment for the virus,” said Stefan Sarafianos, a virologist at Bond LSC and lead author of the study. “Fine-tuned capsid stability is critical for successful infection: too stable a capsid shell and the cargo is never delivered properly; not stable enough and the contents are detected by our immune defenses, triggering an antiviral response. Capsid stability is a key to the puzzle, and to solve it you have to understand its structure.”
This is the most complete model yet of an HIV-1 capsid protein. In a virus, the protein combines in groups of five or six — called pentamers and hexamers, respectively — that assemble into a mosaic that forms the capsid shell. Roughly 1,500 copies of the protein, grouped into about 250 hexamers and 12 pentamers, comprise the capsid.
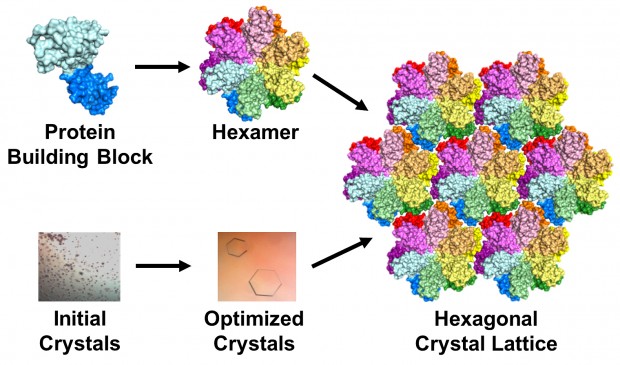
The protein building block of HIV capsid (top left) can assemble to form a hexamer (top middle). Crystals grown using this building block (bottom left and middle) contain an array or lattice of hexamers (right). | Image by Karen Kirby and Anna Gres
HIV, or human immunodeficiency virus, is the retrovirus that leads to AIDS — acquired immunodeficiency syndrome. Roughly 1.2 million people live with HIV in the United States, according to the Centers for Disease Control and Prevention. Globally, about 35 million people were living with HIV in 2013.
A lucky break
Over the years, scientists have employed various techniques and tricks to figure out the structure of the capsid protein. But until now, the clearest image had been made of a mutated version of the protein. It was a compromise: the mutation made the protein stable enough that the scientists could get a good snapshot, but they couldn’t see the detailed interactions between hexamers.
Sarafianos’ lab figured out how to get the full picture: a detailed image of the unmodified proteins that filled all the gaps between hexamers.
The team used a technique called X-ray crystallography to unravel the protein’s secrets. Basically, they took many copies of the protein and coaxed them into forming a patterned, crystalline lattice. Next they shot high-powered X-ray beams at the crystal. By interpreting how the X-rays scattered when they ricocheted off the proteins, the researchers made a 3-D map of the protein.
Karen Kirby
“But it doesn’t make sense until we make an atomic model of the protein to fit in that map,” said Karen Kirby, a research scientist at Bond LSC. “The map is just a grid that you can’t really interpret unless you put a model into it to see ‘Ok, it looks like this part is here, and that part is there, and this is how the protein is put together.’”
The researchers altered, tested and honed their 3-D model until it exactly matched the map produced by the X-ray diffraction pattern. This can be difficult and painstaking, but the researchers’ greatest challenge was creating the protein crystals in the first place: Scientists had been trying to crystallize the unmodified version of the HIV protein for decades without success.
To make a crystal, proteins are suspended in a liquid then slowly precipitated out, just like a “grow your own crystals” kit. But there are a lot of variables that control the process, from salts and additives in the liquid to the amount of protein in the mixture.
“It’s a very delicate balance to grow crystals,” Kirby said. “Many people call it more of an art than a science. It’s frustrating because you can never predict which solution will grow crystals. There are a large number of variables.”
Initially, most arrangements the researchers tried resulted in useless brown junk, Kirby said, caused by the proteins forming solids too quickly. Anna Gres, an MU chemistry grad student who led the project, used a crystallization robot to screen roughly 2,500 conditions.
That was the easy part, Gres said: “The real challenge begins afterwards, as one needs to manually optimize the initial crystallization conditions to find the one that will produce protein crystals of desired quality. This process can take years. In our case, I think we were lucky: It took approximately 500 manual screenings and about 6 month.” But the hard work paid off when she was finally able to produce lovely, hexagonal crystals. Surprisingly, the crystals formed in groups of six proteins, which matched their formation in the viral capsid.
The transition from tiny, useless particulate to invaluable crystals was tremendously exciting, Kirby said. But even to Kirby and Sarafianos, why their attempts succeeded when many others failed remains a little mysterious.
“I still don’t know what are the fine details that made the difference,” Sarafianos said.
“That’s the million dollar question,” said Kirby. “We really don’t have a good answer for that.”
Although solving the enigmatic crystal structure of the native full-length capsid protein was really rewarding, Gres said, she will continue to tinker with her technique: “I am still trying to optimize crystallization conditions, hoping to improve the quality of the crystals and diffraction.”
Water, water everywhere
Once the researchers got a good look at the interactions between hexamers, they were surprised by what they found.
Based on the genetic sequence of the protein, scientists speculated that they would be hydrophobic, or repel water. Instead, they found that “ordered” water molecules at specific sites played a crucial structural role by bridging interactions at the interface between hexamers.
“We thought, ‘How could these lowly waters really be of consequence?’” Sarafianos said. “But if you think about it, there’s 256 of these hexamers in the whole capsid and all kinds of interfaces among them: There’s thousands of water molecules that stabilize the whole structure. We hypothesize that this is an essential part of the stability of the whole capsid molecule.”
To test that hypothesis, they took the crystals, dehydrated them and checked to see if their shape changed. Although the protein lattices may look like sturdy crystals, they’re more like jello, Sarafianos said.
“The protein molecules are precariously touching each other and forming a lattice that is very, very sensitive. It’s held together in this case by water molecules in addition to other interactions.”
The change in shape suggested that water molecules are important in that they allow the capsid to assume different shapes. Moreover, Sarafianos said, the capsid’s malleability and plasticity could be critical to the life cycle of the virus and allow it to act as a multi-functional molecular Swiss army knife.
Onward with research
A clearer image of the capsid protein, could help Sarafianos’ lab gain a better understanding of how the body combats the virus and to discover new ways to disrupt the viral capsid.
“Now we have a system to study effects of capsid-targeting compounds with novel mechanism of action,” Gres said.
Working with a medicinal chemist, Sarafianos’ lab will undertake an iterative process of making compounds, solving their structures, testing them against HIV and then refining the molecules, with the ultimate aim of producing new and effective antiviral drugs.
Endocrine disruptors alter parent behavior in California mice
California mice exposed to bisphenol A (BPA) or ethinyl estradiol changed their parenting behavior, according to an MU Bond LSC study. | Photo by Roger Meissen, Bond LSC
By Roger Meissen | MU Bond Life Sciences Center
What if a chemical changes the way an animal parents?
That could happen due to endocrine disruptors like bisphenol A (BPA).
A recent study of California mice exposed to BPA showed parents spend less time feeding, grooming and interacting with their babies, according to University of Missouri research. Even mother mice not exposed to the chemical parented differently if their male partner was exposed during development.
Most studies only use laboratory mice and rats — where the mother is the sole parental provider — so how early contact to BPA may affect the father and his partner remained a critical gap in existing research.
Bond LSC researcher Cheryl Rosenfeld | Photo by Roger Meissen, Bond LSC
“The nature and extent of care received by an infant is important because it can affect social, emotional and cognitive development,” said Cheryl Rosenfeld, a researcher in MU’s Bond Life Sciences Center and associate professor of biomedical sciences in the College of Veterinary Medicine. “We found that females who were exposed early on to BPA spent less time nursing, so the pups likely did not receive the normal health benefits ascribed to nursing. Likewise, we found that developmental exposure of males and females resulted in them spending more time out of the nest and away from their pups, further suggesting that biparental care was reduced.”
BPA and other endocrine disrupting chemicals like ethinyl estradiol (EE) — found in birth control — concern scientists because they build up in the environment and mimic natural hormones produced by animals, including humans. Everyday exposure to these chemicals can impact offspring development and now have been found to alter adult behavior in test animals.
California mice have special significance for studying parental behavior. Unlike most lab mice, Californian mice pair up to mate and care for offspring. This monogamous behavior could give researchers insight into child rearing behavior found in most human societies and other biparental animals that would be impossible to measure in lab mice and rats.
MU graduate student and primary author Sarah Johnson worked with Rosenfeld to design the study to look at both sexes. Female and male mice were fed one of three diets — food supplemented with BPA or ethinyl estradiol or endocrine-free (control) food — two weeks before breeding. The mice were then randomly paired with the same mate for the entire study. The behavior of both sexes was then tracked for activities like time spent grooming pups, time spent in the nest and time mothers spent nursing.
But how do you measure the behavior of parents?
Rosenfeld’s team depended on hundreds of hours of video footage, taken at particular times of day and night for seven days, starting two days prior to birth. By using infrared cameras they tracked all 56 litters of mice, logging the number of and duration of activities mothers and fathers completed. During this time, the body weight and temperature of the F2 pups, who were not directly or fetally exposed to any chemicals, was logged to monitor their development.
While results showed reduced pup attention from BPA/EE exposed mother mice, the most intriguing result showed that unexposed moms mated with exposed fathers reduced the time they groomed and cared for offspring.
“These female mice have not been exposed here, but if you can see they are still reducing parental care when paired with the BPA/EE-exposed males,” Rosenfeld said. “And what’s even more interesting is that if a mother and father are both exposed, that parental care diminishes further, and becomes even more statistically significant.”
Researchers hope these results will spur others to look at long-term effects of endocrine disruptors on parenting behavior from generation to generation in animal models and, more importantly, in humans, to see if these chemicals can disrupt parental behavior of mothers and fathers, and if so, whether these effects can be transmitted to subsequent generations.